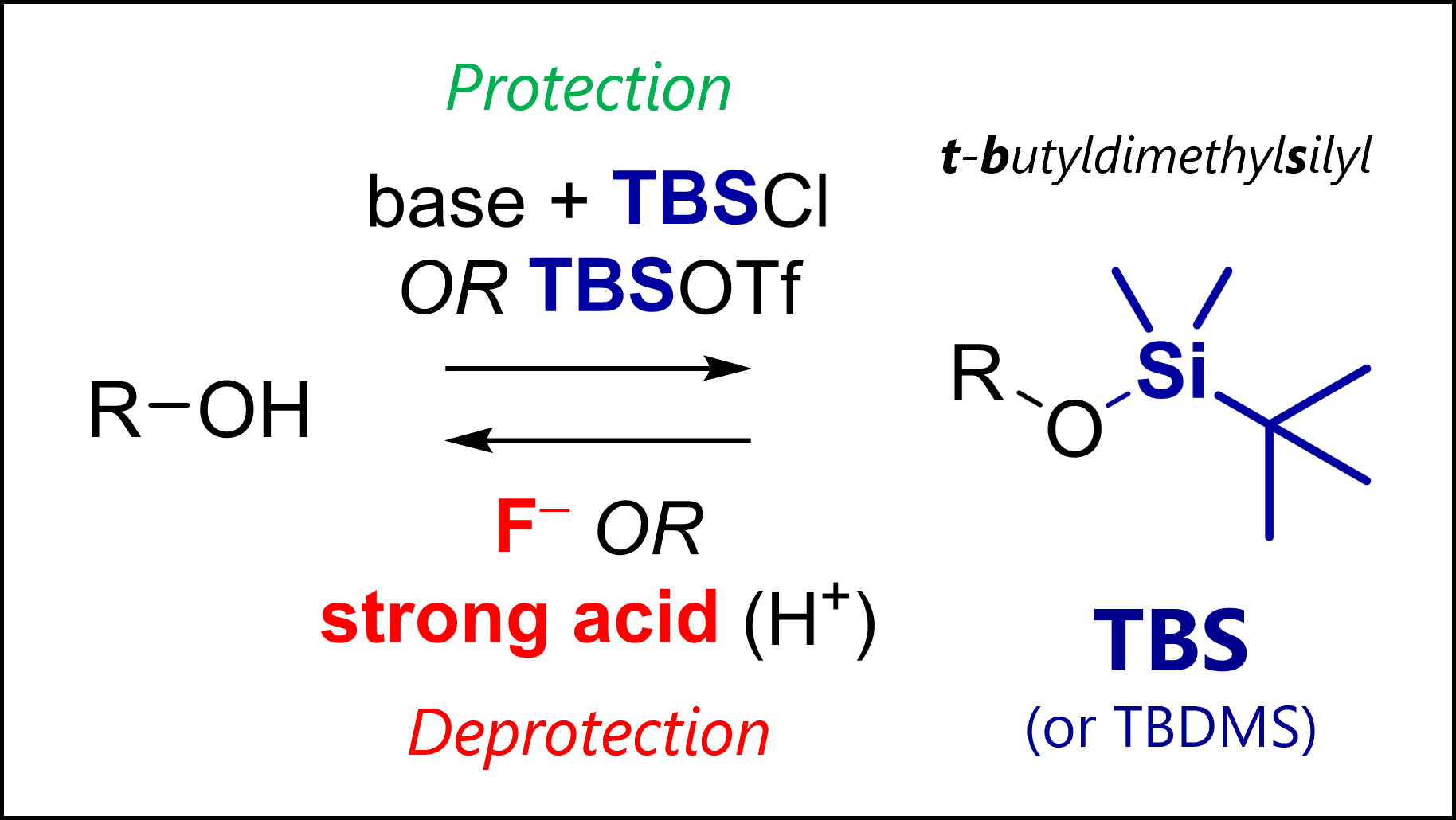
The TBS protecting group protects alcohols in organic synthesis. TBS is deprotected with fluoride anions (e.g., TBAF) or strong acids.
This bulky silyl PG is resilient, allowing for selective/ orthogonal deprotection of similar groups like TMS.
🫡 Here’s what you’ll learn:
Table of contents
👀 Left: 3D model of TBS. Can you differentiate the methyl from the tBu groups?
What is the TBS Protecting Group?
TBS or TBDMS is short for tert–butyldimethylsilyl, a protecting group for alcohols. TBS was introduced by the legendary E. J. Corey in 1972 [1] as an evolution to simpler silyl ethers which had already been known. Note this was right around the time of Fmoc discovery, and relatively late in the history of organic synthesis.
The research already covered protection and deprotection conditions still used today. It is one of the most cited JACS publications ever. A decade later, Corey also introduced triflate reagents for silyl ether synthesis [2].
TBS Protection Mechanism
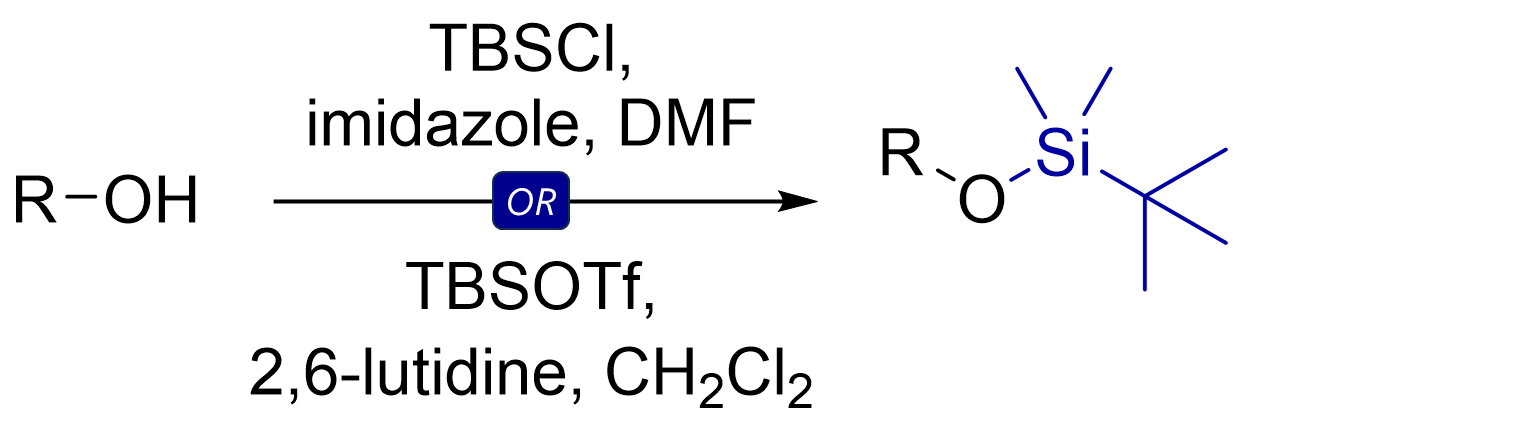
The most common TBS protection conditions are TBS-Cl (forms hygroscopic white crystals) in DMF with imidazole or DMAP. Corey encountered challenges using TBS-Cl for protection of tertiary or hindered secondary alcohols. Use of TBS-OTf triflate (with 2,6-lutidine as base in solvents like dichloromethane) proved more potent for such cases.
The classic mechanism contains generation of an imidazolium intermediate which acts as a silyl transfer reagent for the actual silylation. The steps might look counterintuitive. Silicon, in contrast to carbon, can have five bonds at once! Both silylations likely proceed through associative substitutions with pentavalent silicon intermediates. However, you’re likely also fine drawing a concerted step.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
TBS Deprotection Mechanisms
There are three types of deprotection mechanisms for TBS and silyl ethers in general.
1.Acidic hydrolysis works through protonation of the protected oxygen atom, followed by associate hydrolysis with a pentavalent silicon intermediate.
2.Fluoride-mediated deprotection is based on nucleophilic attack onto silicon, giving a pentacoordinate siliconate intermediate that can release our deprotected product. This is like the imidazole catalysis we’ve seen during TBS protection.
The thermodynamic driving force is the formation of the exceptionally strong Si-F bond (>30 kcal/mol stronger than Si-O) bond. For fluoride-mediated deprotection, TBAF (tetrabutylammonium fluoride) is the archetypical fluoride source. However, other sources like HF or amine-HF complexes are also commonly used.
3.Basic hydrolysis can theoretically also occur with TBS ethers, even though have high stability towards bases. However, at very forcing basic conditions, TBS ethers can hydrolyse (e.g., excess LiOH, dioxane, EtOH, H2O, 90 °C). The mechanism is also based on nucleophilic addition to silicon, followed by Si-O bond breaking.
Silyl ether protecting group Stability
Many different variants of silyl ethers exist. Despite their overall similarity, they can behave quasi-orthogonal due to different lability. Below, you can see the relative stability towards acidic and basic aqueous hydrolysis.
Silyl ether protecting group
Stability to acid
Stability to base
TMS (trimethylsilyl)
1
1
TES (triethylsilyl)
~60
~10-100
TBS (tertbutyldimethylsilyl)
~20’000
~20’000 (likely higher as basic stability > acidic)
TIPS (triisopropylsilyl)
~700’000
~100’000
Stability data from [3]
As you can see, bigger silyl ethers are more stable than smaller ones. This stability directly shows – TBDMS ethers are stable to chromatography and survive various reaction conditions which smaller ethers do not. With different labilities, chemists can deprotect TMS or TES ethers inpresence of TBS protecting groups (more below).
Selective Deprotections of silyl ethers
So, we have mentioned that selective deprotection of silyl ethers might be possible. The first category of reactions follows the steric stability we laid out above. Let’s look at some examples [4].
Pyridinium p-toluene sulfonate (PPTS) in protic solvents like MeOH is the mildest system for deprotection of TBS ethers. In this example, you can see that the conditions are controlled enough so that the bulkier TIPS group does not fall off. The same would go for TBDPS (tert-butyldiphenylsilyl).
The next step in the synthesis is addition of K2CO3 to the diol. What is the product?
This next example is sneaky. Don’t be fooled by seeing “TMS” and thinking it will immediately be less stable than TBS! Upon close inspection, we realize that this is actually a alkyl silyl group, part of the so-called SEM protecting group. It can be removed through fluoride anions or more forcing acidic conditions.
Another question: Propose a mechanism the deprotection of SEM with TBAF!
Welcome to chemistry, it’s sometimes random
The second category of selective reactions does not follow any obvious rules. It’s really experimental randomness. A commonly cited example is from the synthesis of zaragozic acid C [5].
During this effort, it was possible to differentiate between two very similar TBS protecting groups. The use of dichloroacetic acid in MeOH was mild enough to selectively (90% yield) cleave only one of the TBS groups.
That same synthesis actually also nicely demonstrated the concept of selective protection based on steric hindrance. After full consumption and protection of the primary alcohol with TBS-Cl, the chemists threw in TMS-Cl to protect the tertiary hydroxyl group as well.
There’s a recent example (2024) of TBS randomness [6]. In the last synthetic steps towards (+)-heilonine, the authors performed a Clemmensen reduction with Zn in HCl to reduce the ketone. It would have been nice if this one-two punch would have directly delivered the natural product. However, for no apparent reason, only one of the two TBS groups fell off. So, a separate deprotection step with TfOH was required.
“To a solution of the diol (57.48 g, 0.354 mol) in DMF (354 mL) were added imidazole (96.52 g, 1.418 mol) and TBSCl (160.27 g, 1.063 mol) at rt. After stirring at 50 °C for 17h. H2O was added and the mixture was extracted with Et2O. The organic layer was washed with brine, dried over MgSO4, and evaporated in vacuo. The residue was purified by flash column chromatography (SiO2; n-hexane:EtOAc = 10:1) to give the diTBS ether (138.48 g, 100%).”
TBS deprotection experimental procedure [7]
“To a solution of the alcohol (45.2 g) in THF (350 mL) was added TBAF (1.0 M in THF, 184 mL, 0.184 mol) at rt. After stirring at rt for 18 h, the mixture was concentrated in vacuo. The residue was purified by flash column chromatography (SiO2; n-hexane:EtOAc = 4:6→EtOAc) to give diol (29.1 g, 97% yield, 2 steps).”
Do you struggle to comprehend the SN2 mechanism, or the difference between SN2 vs SN1? You are not alone! All of us need models and practice to understand what the molecules look like in their 3D structure. On my channel, you can find some more visual explanations and animations that might help.
SN2 Mechanism: it takes two to tango
Our first model reaction is the nucleophilic substitution of 2-bromobutane with the phenolate anion, also called a Williamson ether synthesis.
2-Bromobutane is chiral as one of the carbons has four different substituents. We are looking at the (R)-enantiomer here – this will be important for the stereospecificity of the reaction. Electrophiles provide the LUMO for reactions, in this case the antibonding sigma star orbital between carbon and our leaving group. Note that bromide and iodide are particularly potent leaving groups due to high acidity of conjugate acids but also weak bonds with carbon. This is due to weak overlap of atomic orbitals, resulting in a low-energy sigma star that is accessible to our nucleophile, phenolate.
This electron-rich anion is completely planar due to conjugation of one of the oxygen electron pairs with the aromatic ring. Its HOMO is localized on the oxygen as you would expect – but we can also nicely see resonance with delocalization across the pi-system.
SN2 Transition state
To ensure decent orbital overlap, the substitution proceeds via back-side attack. Because the nucleophile needs to get pretty close to the already tetrahedral carbon, steric factors are more important for the SN2 reaction compared to SN1.
The SN2 mechanism proceeds in one concerted step with both electrophile and nucleophile present in the transition state – that’s why we call it 2, for bimolecular. The carbon-bromine bond is partially broken, and the carbon-oxygen bond partially formed. Remember that is just a transient energy maximum and not a real intermediate, carbons are never actually five-coordinate!
After the transition state, the product moves to a more comfortable conformation but importantly, features the inverted stereochemistry due to back side attack. This changes the (S) enantiomer in the starting material to the (R) enantiomer product. As a good leaving group, the bromide anion enjoys its solitude and buzzes off.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Steric effects in SN2 substitutions
Due to the five-coordinate transition state, more sterically hindered substrates react much slower or not at all. While it’s not intuitive on paper, the model nicely visualizes that surrounding substituents can block the nucleophiles back side attack. There’s simply too much unwanted repulsion. Instead, depending on reaction conditions like solvent polarity, we would see more step-wise SN1.
Intramolecular SN2 reaction Mechanism
Let’s look at a slightly more advanced example. I’m TotalSynthesis, so I just had to take a cute natural product that was isolated from random tropical algae in Brazil. As fate wanted it, this also has a secondary alkyl bromide, so it fits perfectly.
We’re interested in this epoxide opening step as it showcases a common question on diastereoselectivity. The reaction is intramolecular but is pretty similar to a SN2-type reaction. Given our fixed starting configuration, the side chain and the nucleophilic alkoxide hover on the bottom side of the ring.
As you can see, the nucleophile has a perfect position for the backside attack, leading to the 1,2-anti product. The leaving group is now much worse than bromide, but relieving the strain energy present in the epoxide drives the reaction forward.
Is there another potential substitution? Indeed, the other epoxide carbon is also an electrophile. However, the methyl group at this position adds some steric hindrance. Given the quaternary center, this substitution could also proceed stepwise or “asynchronous”, with C-O bond breaking being more advanced prior to addition.
Taking the longer approach forms a 7-membered ring. Compared to the six-member ring on the right, it’s not as rapidly formed or as stable – but the pathway is still significant with 19% yield.
After six additional reactions, a surprising twist showed that the original proposal was incorrect. It turned out this unique natural product never existed to begin with! Instead, it was a mis-assigned, already known molecule, which is even a bit cooler given it includes two bromides and even a chloride. Well, it happens to the best of us.
I’m looking forward to explaining simple, beginner-level content in addition to my other educational videos. Let me know if this helped you!
The Fmoc protecting group protects amines in organic synthesis. Fmoc is introduced with FmocCl and deprotected with secondary amine bases.
This group is similar to other carbamates (Boc, Cbz) despite being orthogonal. ❓ But there’s more – have you ever heard of Sulfmoc or Bsmoc?
Table of contents
👀 Can you find the acidic C-H bond in this 3D model of Fmoc?
What is the Fmoc Protecting Group?
Fmoc is a fluorenylmethoxycarbonyl group that forms carbamates with amines. However, as a common theme in protecting groups, alcohols and other nucleophiles can also be protected.
Fmoc was introduced by Carpino in 1972 [1]. At that time, few base-sensitive amine protective groups were known. Chemists obviously already used protecting groups, but they were not as straight-forward. For example, the tosylethyloxycarbonyl group (base-labile with KOH/NaOEt) gave the stable carbamate salt which required a second step for decarboxylation.
Fmoc Protection Mechanism
The classic Fmoc protection is with Fmoc-Cl under Schotten-Baumann conditions (e.g. NaHCO3/dioxane/H2O or NaHCO3/DMF), or with anhydrous conditions (e.g. pyridine/CH2Cl2).
If you have seen more than one protecting group, this will not surprise you: The mechanism is attack of the nucleophilic amine to the highly reactive 9-fluorenylmethyl chloroformate. As chloride is the leaving group, the reaction liberates HCl which is neutralized by the base.
Fmoc-Cl can be handled easily (it’s a solid) – however, as it’s an acid chloride, is sensitive to moisture and heat. Thus (as for all protective groups), other “Fmoc+”-equivalent reagents offer more optionality.
Fmoc-OSu is most commonly used nowadays due to the increased stability of this succinimide carbonate. It also has lower unproductive formation of oligopeptides that can occur during preparation of Fmoc amino acid derivatives.
Additional options exist such as Fmoc-OBt or Fmoc-N3. However, you would rather deal with some harmless solids than explosive azides…
Fmoc deprotection mechanism With Base
Fmoc is typically deprotected with secondary amines in DMF. The mechanism has some parallels to Boc. Instead of a stabilized (tertiary) carbocation as an intermediate, Fmoc proceeds through a fluorenyl anion. But why is it stabilized? The position might not seem acidic at first sight.
Upon closer inspection, we see the deprotonated system fulfils Hückel’s rule for n=3 (14 electrons) and is aromatic! That’s why the pKa of fluorenyl is around ~23 (DMSO). This is basically a cyclopentadiene anion (whose aromaticity you will know) sandwiched between two benzene rings.
The intermediary carbanion can eliminate the carbamate in a E1cb mechanism, releasing dibenzofulvene. This side product lead to byproducts (e.g., reaction with nucleophilic amino acid groups) or polymers. This is why secondary amines like piperidine or morpholine are particularly handy!
They hit two birds with one stone. They cleave Fmoc, and also form a stable adduct with the dibenzofulvene. The “secondary” part is quite important as ammonia does not add to the fulvene system [1].
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Fmoc DeProtecTION Speed
There is another reason why piperidine is the most commonly used base to deprotect Fmoc. This table compares half-lives for Fmoc-ValOH in presence of various amine bases in DMF.
Amine base used for Fmoc deprotection
Half life t1/2
20% piperidine
6 seconds
5% piperidine
20 seconds
50% morpholine
1 minute
50% dicyclohexylamine
35 minutes
50% diisopropylethylamine
10 hours
Half-life data from [2]
Piperidine (and morpholine) deprotection is virtually instantaneous on the second-scale. In contrast, secondary or tertiary amines deprotect Fmoc more slowly (hours) and require higher amounts of base. If you wonder, going from DMF to other solvents like DCM reduces the reaction rate.
Fmoc protecting group Orthogonality
Fmoc is very stable towards acid and electrophiles, tolerating reactive reagents like HBr, trifluoroacetic acid, sulfuric acid and thionyl chloride. Thus, its orthogonal to Boc.
However, it is only quasi-orthogonal to Cbz as it undergoes hydrogenolysis as well! It is less reactive than benzyl groups, so selectivity can be achieved. The reduction can occur under traditional (Pd/C, H2) or different transfer catalytic conditions. The final step of the synthesis of Enkephalin was triple-deprotection of O-Bn, CO2-Bn and N-Fmoc.
Question for you: Enkephalin is a human neuropeptide which binds to the body’s opioid receptors. What is the amino acid sequence of the enkephalin form here?
Fmoc Variants
Let us again look at some more advanced concepts. There are Fmoc-related variants that are more base-labile. By attaching electron-withdrawing substituents like sulfonic acid or halides.
What are the effects? Specifically, Sulfmoc increased proton abstraction by a factor 30 in DCM (vs. Fmoc) using 10% morpholine in DCM, or factor ~10 for 10% piperidine [3]. In specific cases, such labile groups might be pretty useful. By the way, Sulfmoc was introduced by Merrifield who won the Nobel Prize for inventing solid-phase peptide synthesis.
Evidently, this comes from acidification of the fluorenyl position. The 2,7-dibromo Fmoc analog has a pKa value of 17.9 or almost 5 units lower than normal Fmoc!
However, there’s an even cooler analog, also published by Carpino called Bsmoc. It can be cleaved under specific conditions which leave normal Fmoc in tact, but typical conditions with piperidine work as well [4].
Another test for your skills: What is the mechanism of N-Bsmoc cleavage with secondary amines? Don’t scroll to the answer below before thinking about it!
Fmoc in Peptide Synthesis
Fmoc was rapidly adopted in modern peptide chemistry [5]. Compared to the established Boc, it was easy to automate: no corrosive TFA is required, and reaction monitoring is easy due to the fluorene by-product (see deprotection). Fmoc SPPS machines were less expensive and avoided use of unpleasant hydrogen fluoride (HF). The conditions themselves were more compatible with modified peptides (e.g., modification with carbohydrates, phosphorylation…).
As another advantage, the Fmoc protecting group enables orthogonal combination of temporary and permanent protecting groups. During Boc SPPS, iterative use of TFA during each cycle leads to deprotection of small amounts of side-chain protecting groups and cleavage of peptide from polymer support.
Bsmoc solution
Let’s conclude with Bsmoc.
The innovative thing is that it functions as a protecting group and scavenger in one! It’s introduction is analogous to normal Fmoc, using the chloroformate or H-hydroxysuccinimide ester. Instead of a deprotonation with piperidine, we have a Michael addition to the conjugated sulfone.
The free carbamate proceeds to decarboxylate as always, but the piperidine stays on the Bsmoc group (thus, it’s a direct scavenger). You might not expect it but it’s very logical: The initial adduct rearranges after some time to the isomer where the double bond is conjugated to the benzene ring.
The ‘quasi-orthogonal’ conditions for Bsmoc-Fmoc are tris(2-aminoethyl)amine as a base. This primary amine cleaves Bsmoc rapidly while keeping Fmoc in tact. On the flip side, use of more hindered bases like diisopropylamine do not react with Bsmoc but cleave the Fmoc group! This is a consequence of steric hindrance slowing down the nucleophilic Michael addition.
D-Threonine (5.00 g, 42.0 mmol) and Fmoc-succinamide (14.9 g, 44.1 mmol) were dissolved in a 2:1 v/v mixture of THF:saturated aqueous NaHCO3 (100 mL). The reaction mixture was stirred at room temperature for 16 h. The reaction was then diluted with water (50 mL) and the pH of the mixture was adjusted to pH 9 via addition of saturated aqueous NaHCO3. The mixture was extracted with diethyl ether (3 x 50 mL) and the aqueous layer was acidified to pH 1 via addition of 1 M HCl. The acidic aqueous mixture was extracted with ethyl acetate (3 x 100 mL) and the combined organic extracts were washed with brine (100 mL), dried over Na2SO4, filtered and concentrated in vacuo to afford crude Fmoc-D-Thr-OH (14.3 g) as a white foam which was deemed to be sufficiently pure and used without further purification.
Fmoc deprotection experimental procedure [7]
In a vial, SM (2043 mg, 2.5mmol) was added and dissolved in 60 mL of acetonitrile. Then, morpholine (647 uL, 7.5mmol) was added while stirring. The reaction was stirred at room temperature for 24 hours, formation of product and full conversion was confirmed by LC-MS. The reaction was quenched by addition of water and extracted with DCM. The organic 9 phases were combined and washed with aqueous LiCl 5%, dried with sodium sulphate and filtered. The solvent was evaporated and crude product the crude product was purified by silica gel flash chromatography (0-5% MeOH in DCM).
The Cbz protecting group protects amines as carbamates in organic synthesis. Cbz is deprotected with hydrogenolysis (H2).
N-Cbz is unique because it’s orthogonal to numerous protecting groups (stable to bases and acids). ➡️ What you’ll learn here:
Table of contents
👀 Left: 3D model of Cbz. Note the orientation of the phenyl ring: it’s not co-planar with the carbamate!
What is the Cbz Protecting Group?
Cbz is a benzyloxycarbonyl group (formerly carboxybenzyl) and protects amines as carbamates. More rarely, chemists might use Cbz to protect alcohols as their carbonates.
Leonidas Zervas (no not the one from the movie “300”) introduced the Cbz group, thus also abbreviated as Z [1]. With this discovery, Leonidas and his advisor Bergmann spearheaded the field of controlled peptide chemistry. In the 1930s, Cbz unlocked the synthesis of previously inaccessible oligopeptides. Zervas continued to be a driving force in peptide chemistry, including development of other protecting groups.
Cbz Protection Mechanism
Cbz protection is typically performed with Cbz-Cl either under Schotten-Baumann conditions (carbonate base) or with an organic base. The mechanism is attack of the nucleophilic amine to the highly reactive chloroformate. As chloride is the leaving group, the reaction liberates HCl and requires some base.
Like we’ve seen for Boc, Cbz2O or other activated agents (e.g., Cbz-OSu) can offer more optionality, depending on the system. If you are into exotic reagents, you might like reagents A or B in the next figure – basically, anything with an activated “Cbz+” synthon works.
As is common the case, we can protect amines selectively given their higher nucleophilicity. However, there have been some reports of challenging selective protection of secondary amines over secondary alcohols. As always, our rules of thumb depend on the specific system at hand.
Cbz deprotection mechanism With Hydrogenolysis
Hydrogenolysis deprotects Cbz protecting groups, usually in an easy and rapid manner. The mechanism is a reduction with H2, releasing toluene and the free carbamate. Consequently, decarboxylation to the deprotected amine is very much favoured (particularly at elevated temperatures).
Besides molecular H2, it is possible to use other “H2 donors” through transfer hydrogenation reactions (e.g., see procedures below).
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Cbz protecting group Orthogonality
Cbz is orthogonal to Boc, Trt, Fmoc and other common protecting groups. As mentioned, it was an essential part of the early days of peptide synthesis.
But beware thinking it’s fully orthogonal! Although Cbz tolerates some acid, harsh conditions can cleave it as well (e.g., excess HCl, HBr)! This mechanism includes protonation of the carbamate and liberation through SN2 and decarboxylation.
Beyond hydrogenation, Cbz can be susceptible to other transition metal catalysis as well, for instance Ni(0) or Pd(0). This interesting case reportdemonstrated selective removal of double Cbz-protected histidine [2] . Compared to heteroaromatic nitrogen atoms, originally basic amines did not engage in any reaction.
This is a great question for you: Suggest a mechanism for the reaction (note the other reagent!) and propose an explanation for the selectivity!
Cbz use cases and tricks
As always, it would be boring to just look at the simplest case of nitrogen protection and deprotection. Let’s briefly discuss three additional topics [3].
First, Cbz protects other nucleophilic functional groups like alcohols, phenols, thiols… as well. In these cases, an organic base (not carbonate) in dichloromethane or some ether-based solvent is typically used. To increase reactivity, NaH as base allows for protection of deactivated, tertiary alcohols:
Notably, Cbz-Cl as a reagent can activate pyridines to regioselective nucleophilic attack. The use of electrophiles for these purposes is a key concept in heterocyclic chemistry. There is a nice collection on 1-acylpyridiniums by the Baran Lab.
Third and coolest, the Cbz group can serve as a masked N-methylamine. Treatment with LiAlH4 exhaustively reduces the carbamate to the alkane.
“To the SM (1.70 g, 2.64 mmol) in THF/H2O (2:1, 15 mL) was added NaHCO3 (443 mg, 5.27 mmol) and Cbz-Cl (0.56 mL, 3.96 mmol) at 0 °C and the solution was stirred for 20 h at the same temperature. The reaction mixture was diluted with H2O and extracted with AcOEt. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The resulting residue was purified by silica gel column chromatography (40% AcOEt/n-hexane) gave 20 (1.85 g, 2.38 mmol) in 90% yield as a white powder.”
Cbz deprotection experimental procedure
[4] Normal hydrogenolysis To a solution of SM (20.7 mg, 15.0 micromol) in 2 mL of MeOH were added 5% Pd-C (6.4 mg), and the mixture was stirred at 60 °C for 40 h under atmospheric pressure of H2. Then, the catalyst was filtered of on pad of celite. The filtrate was concentrated in vacuo to give a crude material containing 27, which was used without purification for the next step.
[5] Deprotection of 5 Cbz groups in the final step via transfer hydrogenation. To a solution of protected caprazamycin A (32) (6.5 mg, 3.21 micromol) in EtOH/HCO2H (1.9 ml, v/v = 20:1) was added Pd black (66.5 mg, 625 micromol) at 25 °C, and the resultant mixture was stirred for 1.5 h at 25 °C. After filtration through celite, the filtrate was concentrated under reduced pressure. The resultant residue was purified by C18 reversed phase. Caprazamycin A eluted at 11-18 min as an isolated peak. The eluent was collected and concentrated under reduced pressure to give a pure caprazamycin A (3.6 mg, 98%) as a colorless powder:
The Boc protecting group protects amines as less reactive carbamates in organic synthesis. Boc is deprotected with acid(H+).
N-Boc teaches us key protecting group concepts and mechanisms. ➡️ What you’ll learn here:
Table of contents
👀 Left: 3D model of Boc
What is the Boc Protecting Group?
Boc stands for tert-butyloxycarbonyl and protects amines as carbamates. More rarely, it is also used to protect alcohols as their carbonates. Boc is resistant to basic hydrolysis, many nucleophiles as well as catalytic hydrogenation. The fact that it can be removed with mild acid makes it orthogonal to other key protecting groups (see below).
Boc Protection Mechanism
The main method of Boc protection is use of Boc anhydride. Base is not strictly needed for the reaction with Boc2O (tert-butanol formed as product). However, bases like triethylamine or NaOH (amino acids) are sometimes used, depending on the system.
The mechanism is straight-forward: attack of the nucleophilic amine to the electrophilic anhydride. The carbonate leaving group can release CO2, providing a strong driving force for the reaction. This step is the same as for other carbamate protecting groups such as Cbz.
As we have seen for PMB, many variants of activated agents can exist. The same is true for Boc. For instance, we can also use Boc-Cl (t-butyl chloroformate) but because it’s unstable and needs to be prepared freshly, Boc2O is much more convenient. Boc-ON is another variant; it’s an “oxyimino-nitrile” reagent.
As a common theme for protecting groups: Remember that amines are more nucleophilicity than alcohols, so you can selectively protect them in many cases.
Boc deprotection mechanism with acid
Acids like TFA, HCl… can deprotect Boc groups. Protonation of the oxygen triggers fragmentation into a stabilized tertiary cation (inductive effect). It later deprotonates to form gaseous isobutene. The fragmented carbamate can decarboxylate, releasing CO2(here you have a parallel of Boc anhydride introduction and its removal) and giving the free amine.
Given the high steric hindrance of the carbamate, the Boc group is not base-labile like methyl esters, for instance.
A potential issue are intermolecular side reactions of the intermediary t-butyl cation. An example from peptide chemistry is alkylation of nucleophilic amino acids methionine or tryptophan. That’s why you might see conditions which employ scavengers like thiophenol, anisole, cresol… to remove the reactive intermediate.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Boc protecting group Orthogonality
Boc protection is a key tool for heterocycle and peptide synthesis. In solid phase peptide synthesis (SPPS), it is used as a protecting group for alpha-amino groups and amino acids lysine, tryptophan and histidine.
Due to its acidic deprotection, it is orthogonal to other important amino acid protecting groups:
Fmoc(9-fluorenylmethoxycarbonyl) – removed with base
Cbz / Z (benzyloxycarbonyl) – removed with H2 reduction
Alloc (allyloxycarbonyl) – removed with transition metal catalysis (Pd)
However, Boc is not stable to Lewis acids or oxidative conditions. We have seen that the O-PMB protecting group is removable with DDQ. In one case [1], treatment of the PMB ether did not result in any desired deprotection.
Here’s some homework: Can you explain why this side reaction and overoxidation might have occurred?
Boc side reactions
In organic chemistry, surprises wait around every corner. Don’t assume that protecting groups stay on forever, and only cleave on demand!
Before, we have mentioned that alcohols are less nucleophilic than amines, allowing selective protections. In this example [2], the authors observed a N-O Boc transfer when preparing a chiral oxazolidinone auxiliary. Upon deprotection of the TBDMS group, the highly nucleophilic alkoxide grabs the Boc group from the nitrogen!
Overman et al used a more conscious Boc-participation in their synthesis towards the diazatricyclic core of sarain marine alkaloids. [3] Base generates the alkoxide which again attacks Boc. This time however, the group is not transferred (the negative charge would not be stabilized on the nitrogen, unlike the previous example). Instead, we see an intramolecular cyclization.
Boc in Peptide Synthesis
As alluded to above – particularly in the early days – the Boc protecting group proved very useful for solid-phase peptide synthesis (SPPS). However, in the late 20th century, the Fmoc protecting group started to replace Boc methodology.
Fmoc deprotection is generally milder than the moderate/ strong acidolysis steps used for Boc. More specifically, Fmoc proved more compatible with synthesis of amino acids that are susceptible to acid-catalysed side reactions.
A good example is the synthesis of the peptide gramicidin A which contains four acid-sensitive tryptophan residues. Using Boc chemistry, yields were in the range of 5-24%. Switching to Fmoc dramatically improved the yields, in some cases to 87% [4].
Closing Remarks
The Boc group is pretty cool, and its deprotection mechanism is a must-know for every organic chemistry student. The orthogonality to base- or reduction-labile protecting groups make it a top pick for many total and peptide syntheses.
However, like all protecting groups, it has its downsides. The carbamate carbon remains somewhat nucleophilic which opens avenues for surprising reactions, particularly intramolecular. Also, the acidic conditions and reactive tert-butyl-cations used can pose challenges to some systems. As always, smart planning and workarounds might be needed.
“2-Bromophenylhydrazine hydrochloride (1.75 g, 7.83 mmol, 1 eq) was dissolved in THF (7.8 mL). Boc anhydride (1.71 g, 7.83 mmol, 1 eq) and NEt3(1.31 ml, 9.40 mmol, 1.2 eq) were added and the reaction mixture was allowed to stir at r.t. overnight. After this time, the reaction mixture was concentrated in vacuo. The crude product was purified by flash column chromatography eluting with 100% hexane to 2% EtOAc in hexane to yield an orange solid (2.41 g, 8.43 mmol, quantitative).”
BOC Protection experimental procedure 2 [6]
One pot Cbz->Boc switch “To a solution of Cbz-carbamate SM (3.5 g, 6.81 mmol) in MeOH (25 mL) were added Pd/C (10 % w/w, 200 mg, 0.19 mmol) and Boc2O (2.17 g, 9.9 mmol) at room temperature. The reaction mixture was stirred under a hydrogen atmosphere (balloon) at room temperature for 6 h. The reaction mixture was filtered through a pad of celite, and then concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc 10:1 → 7:1) afforded Boc-carbamate 42 (2.94 g, 90 %) as an oil.”
BOC deprotection experimental procedure 1 [7]
“Boc-L-allo-End(Cbz)2-OtBu (597 mg, 1 mmol) was dissolved in a mixture of TFA (10 mL) and water (1.0 mL). The mixture was stirred at room temperature for 3 h, then concentrated to give a brown oil. The resulting crude oil was azeotroped with toluene (3 x 10 mL) and concentrated in vacuo to remove any residual TFA.”
BOC deprotection experimental procedure 2 [5]
“Tert-Butyl (R)-(2-(2-(2,5-dichlorophenyl)pyrrolidin-1-yl)-2-oxoethyl)carbamate (0.13 g, 0.33 mmol, 1 eq) was dissolved in a mixture of DCM:TFA (5:1, 8 mL) and the reaction mixture was allowed to stir at room temperature until TLC showed disappearance of starting material. The reaction mixture was concentrated in vacuo to afford the free amine as a trifluoroacetate salt and was used directly without further purification.”
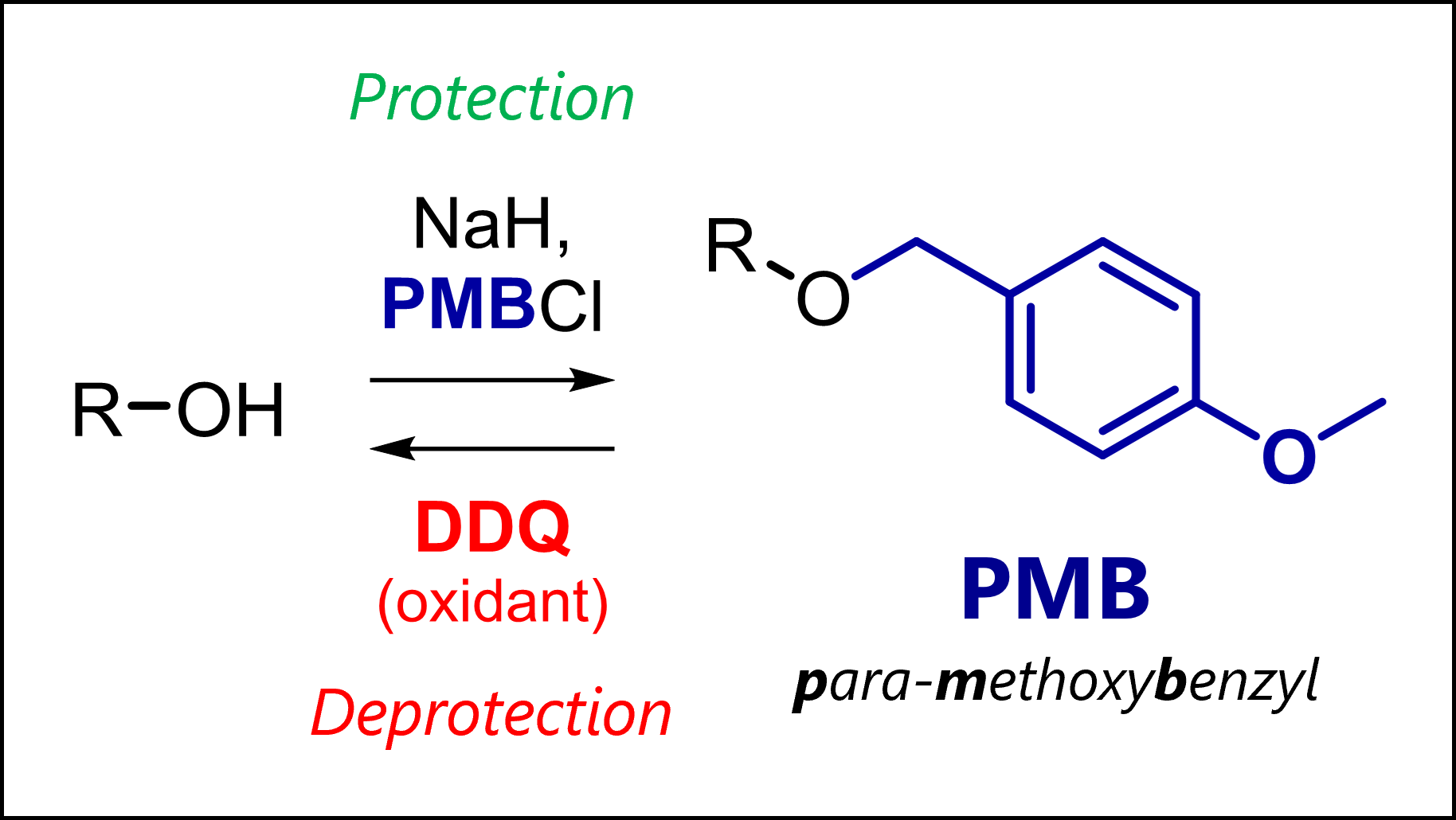
The PMB protecting group protects alcoholsas less reactive ethers in organic synthesis. PMB is deprotected oxidatively or with strong acids.
🫡 Here’s what you’ll learn here:
Table of contents
👀 Left: 3D model of the PMB group to help you visualize it.
What is the PMB Protecting Group?
PMB is a para-methoxybenzyl group, introduced by Yonemitsu in 1982 [1] to protect alcohols and other nucleophilic functional groups. Although PMB ethers are less stable to acid than normal benzyl ethers, they can uniquely be cleaved oxidatively (see below). This allows selective deprotection protocols which becomes critical in complex organic synthesis.
PMB Protection Mechanism
The main method of PMB protection is the Williamson ether synthesis. This reaction uses a moderately strong base to generate an alkoxide which undergoes SN2 substitution with an activated agent like PMB-Cl. Typical conditions include sodium hydride NaH in THF/DMF or DMSO. However, stronger bases like nBuLi work as well.
Beyond PMB-Cl, the reagent you will see most, there are also other methods of PMB protection. Beyond halide variants like PMB-I or PMB-Br, PMB-trichloroacetimidate with catalytic acid can also protect hindered tertiary alcohols. This is due to its higher reactivity. The PMB-pyridyl thiocarbonate with silver(I) is another example.
Interestingly, tetrabutylammonium iodide can be used catalytically for sluggish reactions as well. Here’s a question for you: Do you know how that catalysis works?
PMB DeProtecTION Mechanism WITH DDQ
This protective group differs from others in that it undergoes easy single electron transfer (SET) with DDQ (2,3-dichloro-5,6-dicyano-l,4-benzoquinone). The electron-donating methoxy group stabilizes intermediary radical and oxonium ion. Normal benzyl protecting groups oxidize as well, but much slower than PMB. Obviously, this is due to the O-PMB methoxy group.
After some proton exchanges, water captures the carbocation. As with every redox reaction, the electrons removed from O-PMB (oxidation) end up in the reduced hydroquinone product. Compared to the quinone in DDQ, this system is aromatic.
The hemiacetal formed after water addition can fragment to give the deprotected hydroxy – as well as anisaldehyde. One drawback of is unintended side reaction of the aldehyde or intermediary PMB cations with nucleophilic functional groups, as well as polymerization. Thus, it is common to add nucleophilic scavengers (e.g., thiols) that capture these reactive species. This is a common thread for some deprotections (e.g., Boc).
By the way, other oxidants like cerium(IV) ammonium nitrate (CAN) or NBS might work when DDQ fails.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
PMB protecting group Orthogonality
The DDQ reduction (typically 1.1-1.5 equivalents DDQ in dichloromethane-water mixtures) leaves several functional groups and other protecting groups (MOM, THP, TBS, Bz…) alone. This makes PMB an interesting orthogonal protecting group.
However, electron-rich groups like dienes or trienes can be unintended victims of DDQ. This makes complete sense: electron-rich groups are nucleophiles and thus like to react with oxidants. In some cases, conjugation of dienes with electron-withdrawing groups sufficiently deactivates them, avoiding DDQ interference.
Also, always be on the lookout for unique systems and reactivities! The synthesis of sterepolide, an antibiotic fungal metabolite, exemplifies this concept in oxidative deprotections [2]. When using an excess of DDQ (8 equivalents), the authors found the allylic O-PMB over-oxidized directly to the ketone. In this case, this was convenient as it gave the target natural product, saving one step. However, this can complicate cases where we need deprotection only (most of the time).
Advanced Question: Special PMB Deprotection
As a twist on the previous information, you can ponder on this research [3]. Using 0.5 equivalents of oxalyl chloride led to efficient PMB deprotection of various substrates.
What could be a mechanism for this unique deprotection reaction?
Closing Remarks
PMB is a quite unique protecting group given the ability to remove it oxidatively (in addition to normal acid-mediated cleavage, not explicitly discussed here). In addition, PMB can also protect carboxylic acids, thiols, amines, amides… – or even phosphates! Basically, many other nucleophilic groups. The protection and oxidative removal make it a standard question in organic chemistry courses.
“To an ice-water cooled solution of SM (3.91 g, 15.2 mmol, 1 equiv) in THF-DMF (100 mL-30mL) was added NaH (2.43 g, 60.8 mmol, 4 equiv, 60% suspended in mineral oil) portionwise. After addition, the reaction was stirred at the same temperature until cease of gas releasing, then p-methoxybenzyl bromide (6.11 g, 30.4 mmol, 2 equiv) in THF (25 mL) was slowly added at 0 °C. The reaction mixture was stirred at 0 °C for 1 h, then quenched by slowly adding 1M solution of NaOMe in MeOH (15 mL). The reaction mixture was diluted with EtOAc (300 mL) and washed with water and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The resulting residue was purified by silica gel flash chromatography (hexanes : EtOAc = 25:1-10:1) to give 5.28 g of product as a colorless oil in 92% yield.”
PMB deprotection experimental procedure [5]
“To a solution of SM (1.97 g, 3.95 mmol) in CH2Cl2:0.1 M pH 7 sodium phosphate buffer (18:1, 47 mL) at 0 °C was added 2,3-dichloro-5,6-dicyano-p-benzoquinone (1.17 g, 5.14 mmol) slowly as a solid. The reaction was warmed to rt and stirred for 1 h. The crude mixture was directly loaded onto a silica gel column with a top layer of MgSO4:sand (1:1, 0.5 inches). Elution with 5% to 30% EtOAc in hexanes yielded the product (1.45 g, 97%).”
In this post, we will look at MDMA’s history and its chemical syntheses. We will dispel myths about MDMA’s discovery and review the first kilo-gram scale MDMA synthesis published in a journal. We also dissect impressive recent clinical data that suggest ecstasy might help up to millions of people affected by PTSD. This might not be surprising if you’ve seen our discussion of psilocybin, ibogaine or LSD.
How to Make MDMA?
I’m not sorry to disappoint you. This is purely educational and not a DIY-tutorial for illegal clandestine synthesis. Also, don’t use this information to rationalize your drug use. Long-term and side effects of MDMA can be severe and regular use is discouraged even by clinical advocates.
MDMA history
The origin of MDMA has quite some tales associated with it. For example, crediting various German scientists with its discovery, even though no documentation or basis for this can be found. MDMA also was not intended for use in World War 1. However, there was quite some military experimentation on stimulants later on in the 1950s. The first point at least goes in the right direction, but the history is much more intriguing than this.
The story actually starts with hydrastine, an anti-hemorrhagic natural product isolated from some random plant. By the 20th century, this drug became more expensive because the plant was becoming rarer and cultivation attempts failed. Therefore, the German company Merck was interested in finding ways to chemically synthesize it. They had a chad chemist reach out and offer a new, cheap synthetic procedure for hydrastine. For some reason, this guy signed the contract with Merck’s competitor Bayer which is quite funny. So the Merck scientists now had to find some new anti-hemorrhagic agents or new syntheses.
You can appreciate that hydrastine is basically a more beefed up version of MDMA. Not too shockingly, the Merck scientists produced MDMA as a side product, and were not interested in it at all. While their 1912 patent refers to MDMA’s structure for the first time, they did not pursue or test it. Thus, the first MDMA publication and synthesis was published only 50 years later. Things gained traction from there on.
MDMA Synthesis from Safrole
Let’s check out three syntheses of MDMA starting with Merck’s synthesis from 1912. Second, we will review a late 20th century approach and third, look at the 2022 kilo-scale MDMA synthesis. There are other clandestine methods, actually mentioned in quite a few papers, but obviously we will not discuss this here.
So safrole is a natural product used in the first half of the 20th century as a food flavor. 50 Cent would likely agree, it has a nice candy shop aroma. Human consumption was banned after people realized it increases rates of liver cancer. Feels like half of pesticides and food ingredients have the same story… Safrole was the starting material for Merck, but it can also be made synthetically in a few steps. Starting from Catechol, a double SN2 reaction forms 1,3-benzodioxole. Then, mono-bromination with NBS gives the aryl-bromide. Treatment with magnesium converts into a a Grignard reagent and used in a nucleophilic substitution with allyl bromide.
From safrole it’s only two steps: first, a normal Markovnikov-selective hydrobromination, and another SN2 with methylamine to get MDMA. Optionally, you can also throw in a Finkelstein halogen exchange to get better yields in the substitution.
MDMA Synthesis from Piperonal
The second synthesis from piperonal starts with a Henri condensation reaction, creating a nitro-olefin. This can be reduced in acidic conditions to the ketone and a reductive amination with methylamine gives MDMA. So this synthesis uses a bit less bromines but more redox chemistry.
Large Scale Synthesis of MDMA
The final synthesis is pretty sweet. It was published in 2022 by the MAPS PBC. This is a biopharma company and subsidiary of MAPS, a non-profit working to raise awareness and understanding of psychedelic substances. They required large amounts of MDMA to supply their two Phase 3 clinical trials, which we will check out shortly. This is the first-ever document kilogram scale preparation of ecstasy. The product is appropriate for clinical and potential licensed therapeutic use due to the process’ validation and GMP compliance.
Safrole and piperonal are controlled substances and thus highly regulated and difficult to obtain. Instead, the chemists used an arylbromide (an intermediate towards safrole) that is commercially accessible. This synthesis is similar to others we saw but comes with a twist. It starts again with a Grignard reaction but this time, with 1,2-propylene oxide as an electrophile. This epoxide nicely introduces the rest of the aliphatic chain, leaving a secondary alcohol which can, similar to other syntheses, be oxidized to the ketone. This ketone could be used without any purification in the final reductive amination step. You can check out the paper for more info – they go into some more details on validation and impurities. The experimental procedures are quite funny to read, as they ultimately isolate 3.6kg of MDMA HCl salt with over 99.4% purity.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
PTSD Disease Burden
So they put in a lot of effort in this process – but why is it worthwhile to look at PTSD? As crazy as it sounds, 6-7% of people in the US experience PTSD at some point in their lives, with about 1/3 of cases classified as severe. Often, there are other conditions decreasing chances of successful therapy, so these high-risk patients need more effective treatments. Just as a side note, this did remind me of other shockingly high estimates from the US National Institute of Mental Health – for example, they also state that 19% of adults experience what they termed “any anxiety disorder” per year. This is probably exaggerated, of course anxiety is human but proper clinical disorders are probably not affecting 20% of adults every year.
As a last reason, many patients do not respond to first-line treatment with SSRIs – most notably, those are sertraline and paroxetine. The latter was actually part of the massive $3bn fraud settlement due to unlawful promotion and failure to report safety data. You might know that SSRIs are used in various depressive and anxiety disorders, so it would be nice to have a more targeted therapy or intervention. That’s why MAPS has been supporting MDMA clinical trials as early as 1992. All their advocacy and support culminated in two large-scale Phase 3 trials which were recently completed – we will dissect one of them.
MDMA-Assisted Therapy for PTSD
Let’s talk about study design before going into results – after an initial wash-out of any other psychiatric medications, patients went through four blocks consisting of various therapy sessions. The important points are the red experimental sessions – corresponding to the three occasions where patients in the treatment arm received an 80-120mg dose of MDMA. The individual therapy sessions consisted of supported introspection, experience sharing and probably some other things, and were conducted by trained clinical teams.
This was a placebo-controlled Phase 3 study, so the total 90 patients were randomized to two trial arms. You can see that the patients in each trial had quite comparable characteristics, which obviously is important if you want to compare the effect of a medication – for example, the average duration of PTSD was around 13-15 years for both segments, although there was quite a large variation. From a trial endpoint perspective, there are two important measurements to look at. The CAPS-5 score is based on a semi-quantitative questionnaire that sheds some light on how bad the PTSD is – a score in the 40s, as present in the trial baseline, means very severe PTSD. The Beck Depression Inventory score tells you how depressed someone is – here a score above 30 is also severe.
MDMA-Assisted Therapy for PTSD
How did these severely affected patients they respond to MDMA-supported therapy? Both PTSD severity and depression scores decreased significantly from baseline until end of the last therapy block. You can see that normal therapy also improves outcomes, so these seemingly fluffy therapy sessions are useful – but the effect with MDMA on top is clearly higher. At the end of therapy, patients in the treatment arm were much better off (only mild to moderate PTSD, lower depressive symptoms). Please note that guided therapy was still needed, so just taking MDMA wouldn’t have the same effect and could make it even worse.
While there were quite a few non-responders and only few patients in remission for placebo with therapy, the MDMA group had almost 40% of people completely PTSD-free and only few not responding at all. The nice thing was also that MDMA had an equally positive effect in high-risk people with other disorders, including the especially difficult-to-treat dissociative subtype of PTSD.
Last, MDMA had a quite good safety profile. Side effects like muscle tightness or appetite loss were more frequent in the treatment arm but most are harmless. I would guess that you would rather lose appetite and have some tight muscles, than be afflicted with severe PTSD. More severe adverse events, like suicide attempts or self-harm were actually only observed in the placebo control, probably because their intervention was less effective. So at least in the short-term, there were no concerning safety signals.
It is still a mystery how this works physiologically, but the literature speculates MDMA might reopen a window of neuroplasticity that allows for processing and release of fear and other emotions. Doing so, MDMA might support and catalyze therapeutic processing by allowing patients to stay emotionally engaged while revisiting traumatic experiences without becoming overwhelmed.
MDMA FDA approval in 2024?
The FDA already granted MDMA-assisted therapy a break-through designation in 2017 – so with this promising data in hand, MAPS PBC is expecting to file for FDA approval in late 2023. It will be interesting to see how they decide on this. Let me surprise you with another score which I intentionally left out earlier for simplicity, the Sheehan Disability Scale. This is measures how well an individual functions in key life dimensions, and it seems like MDMA-assisted therapy could also help thousands or millions of people become more functional and independent in their daily lives. Supposedly, US veterans report service-related disabilities that cost the government $73 billion per year. A sizeable chunk of these costs are probably due to PTSD, which might also encourage the FDA to approve MDMA-assisted therapy, at least for high risk patients.
I think this was quite a nice journey, going from almost ancient chemistry to modern clinical outcomes. Thanks for reading and until next time!
This educational article covers a published synthesis of lysergic acid, the precursor of the psychoactive drug lysergic acid diethylamide or LSD.
A team of chemists recently reported a synthesis of LSD in only 6 laboratory steps! We will look at the chemistry behind it and uncover some other insights – for example, how do chemists measure how trippy a molecule is?
Rationale for LSD synthesis
So these scientists, are they a bunch of Breaking Bad wannabes or why would they investigate even more chemical syntheses of LSD? Well, LSD derivatives such as bromocryptine can be pharmacologically useful for treatment of neurological, metabolic and other disorders. This means we want to get more efficient at making LSD-like scaffolds for drug discovery.
In 2020, there was an interesting structure-activity relationship study. It showed for the first time that psychedelic compounds, such as derivatives of DMT, can be engineered lose hallucinogenic side effects while retaining their useful psychoplastogenic properties. The left-hand side 5-methoxy-DMT makes you trip. The isomer with the methoxy substituent shifted by just one carbon, does not. While this might be disappointing for some of you, it’s obviously better if patients are not hallucinating weird shit after taking their pills.
If you wondered – trippy-ness can be estimated by looking at how often mice violently shake their head after administration of psychoactive drugs. This is a well-validated proxy for hallucinations and was first established already 70 years ago! You can see that while 5-methoxy-DMT leads to head twitching, the 6-methoxy isomer has no significant hallucinogenic activity. There’s actually a nice concentration-dependent relationship.
Six-Step Synthesis of Lysergic Acid
So how does this super-quick route look like? This synthesis builds on a key intramolecular Heck reaction which creates the key vinyl bond that is present in LSD. This Heck-approach is not an invention of the 2023 synthesis, as it had been used in previous, longer syntheses already. However, this route efficiently traced the intermediate back to this indole containing. This starting material can be bought commercially and conveniently has the bromo group for the Heck reaction. Obviously this makes a lot more sense than unnecessarily taking apart the indole ring. Let’s take a closer look at the specifics of this synthesis.
The first step was a magnesium-halogen exchange of this iodopyridine to create a heterocyclic nucleophile. This one is happy to attack the electrophilic carbon of the functionalized aldehyde, leaving a hydroxyl group in the product. As you might remember, there is no oxygen in LSDat this position. Thus, the next step simply removed this group by reduction with triethylsilane.
The acid used in this step removed the N-Boc protecting group, so they re-installed afterwards. After this protection, the most nucleophilic group is the pyridine nitrogen – so it was methylated with methyl triflate. This gave a pyridinium salt which was reduced by sodium borohydride. Two hydride equivalents attack the ring: The first one gives the reduced tertiary amine that is part of LSD. The second hydride reduces one of the double bonds, leaving the alpha-beta unsaturated ester. All of this happened in the same reaction vessel. But still, the authors were a bit sneaky to categorize this as just one single step.
But wait – to enable the key Heck coupling reaction, the olefin actually needs to be located at the other carbon. They achieved this by using LiTMP as a strong base. The resulting isomerized anion which can be protonated in a diastereoselective manner. The desired isomer has the ester on the same side as the existing hydrogen of the 6-membered ring. While the preference isn’t great, they formed it in slight excess over the undesired one. Conveniently, they found subjecting it to the same conditions recycled some of it to the desired product.
The Heck reaction proceeded with the standard mechanism. Oxidative addition of Pd(0) allowed for olefin insertion and creation of the C-C bond in blue. Now, given there are two beta-hydrogens available, there are two pathways towards elimination. There’s the orange hydride elimination, and the pink one, which is preferred in a rough 1 to 3 ratio. Note that the stereochemistry of the ester became wobbly again the orange product. This is because the reaction occurred at a 100 degrees with mild base with some isomerization taking place.
Even though we end up with three different products, it’s no big deal. They simply added potassium hydroxide to all, and heated things up to get to lysergic acid in around 50% yield. This is double-deprotection and isomerization. Natural products are usually stable isomers so it’s not surprising that the isomerization forms the configuration present in LSD preferentially. Unfortunately, their final product is not so satisfying as they only isolated a brown solid. I don’t suggest supplying this to the dangerous dealer in the neighborhood. I’ve seen some procedures getting nice white crystals but these folks didn’t care too much about ultra-pure product.
Lastly, they showed that this synthetic route could be useful to explore and study LSD analogs – remember the methoxy-substituted DMT structures at the start? The started with a chloro-substituted indole starting material and replicated the reactions – including the Heck reaction – to create a C12-cholor-lysergic acid derivative. Theoretically, you could create different LSD analogs now by functionalizing the aryl chloride – which might help scientists find future drugs based on LSD with differentiated therapeutic profiles.
The thalidomide tragedy was the biggest “man-made disaster apart from war”.
This molecule’s structure looks simple and innocent, but a chiral carbon gives rise to two enantiomers with different pharmacological effects. While one isomer was a safe sedative, the other one led to limb malformations in thousands of babies. We will check out how this works exactly, but notably, the enantiomers interconvert in the body so there’s no way to control them.
Get this: Despite increasing doubts of its safety, the US distributor of this horror drug aggressively pushed for its approval simply to maximize sales over Christmas!
If you think the heart-breaking tragedy sealed thalidomide’s fate, you couldn’t be more wrong. Half a century after the tragedy, it generated up to $500million sales for a pharma company! But wait, there’s more. A slightly modified, much more expensive version of thalidomide brought in eye-watering 12 billion dollars.
How is it possible that the same drug which inflicted so much damage revived and even influenced one of the biggest acquisitions in pharma history? As we will see, the answer is glue – no joke. The story of course wouldn’t be complete without classic big pharma monopoly games and price hiking. And let’s not forget everyone’s favourites – lawsuits.
In this post, we will go through history, biology, organic chemistry and pharmacology of thalidomide and its many cousins. You will also appreciate some paradoxical and morally questionable aspects of drug development.
What is Thalidomide?
Let’s get into it. Thalidomide has a very simple chemical structure. In the 1950s, the relatively small and inexperienced pharmaceutical company Chemie Grünenthal looked for new antibiotics. Instead of antibiotic activity, thalidomide seemed to be a great sedative and help with sleep or nausea. Unfortunately, it doesn’t help the story that some of Grünenthal’s leaders were ex-Nazi scientist. The research head Mückter was involved in death camp experiments and made bank during his tenure. It started all rosy. Initial safety tests in mice and rats showed good tolerability and no side effects even at remarkably high doses. Back then, you didn’t have to understand how drugs worked at all. So, thalidomide was dubbed completely safe and aggressively marketed in 1957.
It quickly became Germany’s second best-selling pharmaceutical, just behind Bayer’s Aspirin.Ironically,safety was one of its key marketing messages. Many pregnant women used thalidomide for morning sickness. The lack of toxicity was convenient as unlike barbiturates, this agent couldn’t be misused for suicide attempts. However, over the next two years, sudden increases in cases of usually rare limb defects were detected in newborns.
It’s less known that there were prettyearly findings of teratogenicity and neurotoxicity from various researchers. Some of them, like these observations on tad poles, were shared with Grünenthal already in 1959 – with no response.
Thalidomide Side Effects: Beginnings
Because the incidence of deformations increased so unprecedently, German paediatricians suspected an environmental factor. In late 1961, thalidomide use by mothers during early pregnancy was the common factor. Only later did we find out that deformations were actually just the tip of the iceberg. Thalidomide induced many more miscarriages and less obvious defects like organ problems. After increasing noise on the issue, the German government pulled the drug off the market against the company’s wishes. The adverse impact of early use is so high that even a single tablet was enough to induce pregnancy loss or abnormalities – but why? To understand, we need to recap two topics.
First, we need to know about the ubiquitin proteasome system, basically, cellular garbage management. Some of you might remember the process from your biochemistry classes.
The process starts with very few, so-called E1 enzymes which are activated with ubiquitin, a small regulatory protein consisting of roughly 80 amino acids. This green ubiquitin tag is ultimately what directs the degradation of target proteins. As you can imagine, there are thousands of proteins that the cell might want to be able to degrade. However, it would be challenging to do this specifically if all you have are a few different E1 enzymes. This is ubiquitin groups are cascaded to a broader variety of E2 conjugation enzymes which finally put the tag on more than a thousand so called E3 ligases. These enzymes recognize specific substrates and upon addition of enough ubiquitin tags, the proteasome shreds up their targets into smaller peptides.
Second, we need to know about molecular glues. These work exactly like you would think. The glue molecules bind between two different proteins, aggregating or gluing them together. This can lead to several effects, but targeted protein degradation is the most important one.
How does this relate to thalidomide? Well, the molecule binds to the protein cereblon which is part of a E3 ubiquitin ligase complex. Once bound, it can act as a glue between cereblon and neo-substrates, innocent molecules. This exposes them to the E3 ligase machinery, so they are ubiquitinylated and degraded.
Remember that the adhesion arises from nuanced interactions between functional groups of the molecular glue, cereblon in purple, and its neo-substrate in green. We don’t know the natural targets of cereblon but amongst others, it’s critical for brain development, hence its name.
Thalidomide Side Effects: Cereblon
By acting via cereblon, Thalidomide-initiated protein degradation influences the body’s immune system (immunomodulatory drug). The mechanism is very complex, but one of the innocent casualties is SALL4. This is an important transcription factor that governs gene expression for normal limb development. Its absence results in deformations which is why thalidomide proved so dangerous for pregnant women. Actually, genetic deletion of SALL4 replicates a similar phenotype. But why was this missed by Grünenthal? A critical piece of information that was not known in the 1950s – rodents are resistant to thalidomide’s teratogenicity. This explains the absence of safety signals, even at high doses. Only later did people figure out that rabbits or chickens are more sensitive animal models. Why?
The susceptibility comes from a single amino acid difference in cereblon sequences. Primates and rabbits with a valine suffer thalidomide embryopathies – while rodents with an isoleucine did not show any safety signals. Interestingly, the bushbaby bears an isoleucine and is the only known resistant primate. Scientists demonstrated that mutant mice with an unnatural valine at the position become sensitive. Compared to wildtype mice, they showed statistically significant higher miscarriage rates. This is really fascinating – a slightly bulkier amino acid influences binding and degradation of substrates such as CK1 alpha but potentially also SALL4 or others.
If you had a chemistry class or two, you would know that by having one chiral center, thalidomide has two enantiomers. Much too late, it turned out that the (S) enantiomer is ten-fold more potent protein degrader. Giving only the safe isomer as a drug is not an option. The acidic proton at the chiral center triggers partial conversion to the bad enantiomer at pH levels of over 6. If you’ve watched my deuterium video, you will know that this interconversion is slower for deuterium due to its kinetic isotope effect.
How can the almost identical (R)-enantiomer be safe? Although it has the same molecular contacts with cereblon, its affinity is much lower due to an energetically unfavourable conformation that it needs to adopt upon binding. This twisting occurs because the glutarimide ring wants to minimize steric clashing with the binding pocket, particularly the highlighted tryptophan 383.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Thalidomide Synthesis & AFtermath
If we look at its synthesis, it becomes obvious why original thalidomide is a racemic mixture. The original Grünenthal synthesis starts with a condensation of L-glutamic acid and phthalic anhydride. Even though the amino acid used was chiral, the basic conditions and high temperature result in a racemic product due to the. To close the ring, the free acids linked via activation with acetic anhydride and a last treatment with urea introduced the nitrogen.
So, the aftermath entailed a large criminal trial, examining potential negligent behaviour by leading Grünenthal employees. The process was extremely drawn out, probably the dream of every lawyer. 600thousand pages of documents without any clear verdict. Ultimately, it was said that based on the state of science at the time, the teratogenic effects of thalidomide could not have been anticipated – so the trial was terminated and settled between Grünenthal and impacted parents. The company is still providing support to affected persons through a novel foundation, with more than $100 million Euros contributed to date.
Thalidomide in the United States
The impact in the US is another ridiculous story. Grünenthal had offered the company Smith, Kline & French – today’s GSK – to market the drug in North America. SKF ran a large clinical trial which likely also resulted in several phocomelia cases, and they declined Grünenthal’s partnership offer.
However, another company Richardson-Merrell was eager to introduce it. These guys were calling up the FDA to submit a marketing authorization application in the fall of 1960. I invite you to read this nice article which contains comments of Frances Kelsey who reviewed the application at the FDA. And yes, that’s her with President John F. Kennedy, getting a medal for Distinguished Federal Civilian Service. You see, as if this story couldn’t get worse and more capitalistic, Richardson-Merrell was pushing for an early approval prior to Christmas to maximize their sales.
In a – now recognized as heroic – move Kelsey challenged the drug’s data. Something felt off about giving enormous amounts without any toxicity – so suspicions rose that other conditions could change the drug’s absorption and unveil toxic effects. Her suspicions proved to be right – but that didn’t prevent Richardson-Merrell from giving away literally millions of thalidomide tablets for “investigational use”, at the time permissible under existing regulations. The FDA cited 17 children born in America with thalidomide-associated deformities, but the true number is surely higher.
What has science learned from this tragedy? On one hand, drug controls got stricter. Prior to 1962, drug developers only had to show that new drugs were safe – and as we just saw, even that was not a given. A new pivotal amendment required strict “proof of efficacy” from well-controlled studies, and not the bro-science which Richardson-Merrell tried to pass. Drug advertising now required accurate information about side effects, and clinical trials had to include informed consent of participants prior to the study. For us in the 21st century, this seems obvious. The FDA also launched a comprehensive assessment of drugs that were already on the market. Finally, drug testing got more robust with a requirement to use rabbits and other thalidomide-sensitive species for teratogenicity testing.
Is Thalidomide Still Used Today?
Thalidomide’s risks but also benefits continue to linger. Just shortly after its initial withdrawal, it proved efficacious in ENL, a leprosy complication. To avoid teratogenicity, access to the drug depends on so-called Risk Evaluation and Mitigation Strategies, short REMS. For instance, female patients must avoid pregnancy though regular testing and use of two or more forms of reliable contraception. In the US, thalidomide was approved for leprosy in 1998 and REMS were well regulated. However, use over decades in countries like Brazil with subpar REMS has still led to some cases of embryopathy.
Although there was research on thalidomide in cancer already in the 60s, the molecule was finally proven to have anti-cancer activity in the 90s. This time around, the company Celgene got IP rights to the drug and thoroughly interrogated its potential. A landmark trial showcased its value in multiple myeloma, a type of blood cancer. Given its unique mechanism and profile, combination with other agents was powerful. This resurrected thalidomide, turning a monster drug into a precious option for patients who relapsed or did not respond to other treatment.
Mechanistically, the anti-oncology effect arises from degradation of Ikaros and Aiolos. Unlike SALL4, these transcription factors regulate the development of B and T cells of the immune system. Ultimately, thalidomide inhibits the process of angiogenesis. As a very smart person, you will realize that lower growth of new blood vessels in turn suppresses tumor growth.
The “new early” days of thalidomide remained controversial, with a whistleblower lawsuit accusing Celgene of off-label marketing. Allegedly, they actively pushed thalidomide, which as we saw was approved for leprosy, to be off label prescribed to cancer patients prior to its approval. While these off-label prescriptions extended thousands of lives, intentional off-label marketing by companies is not compliant. The company ultimately had to settle the lawsuit for 280 million dollars.
Lenalidomide: multiple myelomA Blockbuster
Thalidomide’s legacy is even more shocking. We already know that small changes have big impacts so you shouldn’t be shocked. You see, two simple functional group modifications created lenalidomide. This is the big boy. By the way, there’s even another hybrid between the two – a bit less imaginative. Due to the new structures, these analogues enjoyed new marketing exclusivity, with great commercial success.
Lenalidomide received an orphan drug designation – a FDA incentive that gives drug developers special tax incentives and market exclusivity. With a hefty original 6-figure price tag, the drug earned Celgene double digit billions in yearly sales.
Lenalidomide didn’t sell for no reason. Molecularly, it is much more potent than thalidomide across various metrics. For instance, its IC50 inhibitory value against resistant multiple myeloma cells is orders of magnitude lower.
These molecular changes also translate into better survival outcomes for patients than thalidomide. In addition, risks of neuropathies and other adverse events were lower. This is a follow-on strategy gone well: new molecule, better efficacy and financial success.
Legal Considerations
You could also argue that given Celgene’s research is not associated with Grünenthal’s initial wrongdoings, they actually changed the world for the better. You could also call it a perverse twist – a horror drug ended up as the basis for massive profit. Some have accused Celgene of using particularly fierce ways of preventing entry of generics. Doing so, it managed to command soaring prices for the drugs, and even increase them. This includes more than a dozen of patents on their REMS system which further blocked generic competition. Remember, REMS are special activities that patients, providers and distributors need to take to prevent harm from teratogenicity.
In this case, it means that access to the drug is dependent on several criteria, such as regular pregnancy tests and surveys. This is great because it encourages safe use of the medicine, but as always, we have more potential illegal activities looming.
For many years, Celgene fought it out with the generics manufacturer Mylan. You see, to call approve a generic medicine, the FDA needs to see bioequivalence data. Essentially, we need to undoubtedly prove that the copy has the same effects. So, generic manufacturers need to buy the branded drug. Allegedly, Celgene not only refused to sell the drugs directly, but they also implemented distribution restrictions that prevented Mylan from buying thalidomide and lenalidomide. This was resolved after five years in classic fashion by paying 100 million dollars and some change to settle the claims.
As alluded to, subsequently BMS acquired Celgene and with it, the knowledge on thalidomide and friends. If you thought this was the end of the saga, think again.
Outlook:More Cereblon Modulators
The development of next-generation cereblon modulators such as iberdomide is still ongoing. At a first glance, this one might look like a simple copycat molecule.
As you can see from the crystal structure, the morpholino side chain extends into a pocket on cereblon, increasing surface interactions and binding of the molecular glue.
This enhanced affinity results in a faster protein degradation of neo-substrates, and anti-proliferative activity against multiple myeloma cell lines. In normal cells, iberdomide is much more potent – compare the red and green curves. More importantly, this analog retains activity in cells resistant to lenalidomide. Due to this higher penetration, the hope is that the drug will prove more efficacious in resistant cases. It’s currently in phase 3 trials and again, the idea is combining it with other medications to stack up the effects.
We will close with a final next-generation idea: covalent modification of cereblon. Scientists have noted that there’s a histidine residue close to the molecular glue binding site. Do you already know where this is heading? By creating analogs with electrophilic groups such as this highly reactive fluorosulfate, we can trigger a covalent bond formation with the proximal histidine.
Why is this even interesting? Well, this covalent modification triggers broader conformational changes which change cereblon’s activity. The scientists found that this covalent modulator led to the degradation of the so far elusive protein NTAQ1. Thus, such experiments might unlock even more avenues for drug discovery in different tumor types.
To not overdo it, we’ll wrap up here. As always, until next time!
Key references on thalidomide science and other information:
Frances Oldham Kelsey. FDA medical reviewer leaves her mark on history | FDA Consum 2001, 35, 24
The Thalidomide Syndrome | Scientific American 1962, 207, 29
THALIDOMIDE AND CONGENITAL ABNORMALITIES | Lancet 1962, 279, 45
The Ubiquitin Proteasome System in Neuromuscular Disorders: Moving Beyond Movement | Int J Mol Sci 2020, 21, 6429
Molecular glues modulate protein functions by inducing protein aggregation: A promising therapeutic strategy of small molecules for disease treatment| Acta Pharmaceutica Sinica B 2022, 12, 3548
Exploiting ubiquitin ligase cereblon as a target for small-molecule compounds in medicine and chemical biology | Cell Chem Biol 2021, 28, 987
Crbn I391V is sufficient to confer in vivo sensitivity to thalidomide and its derivatives in mice | Blood 2018, 132, 1535
Differentiation of antiinflammatory and antitumorigenic properties of stabilized enantiomers of thalidomide analogs | PNAS 2015, E1471
Lawsuit Blames Thalidomide for More Birth Defects | Scientific American 2011
Antitumor Activity of Thalidomide in Refractory Multiple Myeloma | NEJM 1999, 341, 1565
Immunomodulatory Drugs for the Treatment of B Cell Malignancies | Int. J. Mol. Sci. 2021, 22(16), 8572
A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos | J Med Chem 2018, 61, 535
This post explains the question: What is total synthesis? If you read this, you might be forced to study organic chemistry 🙁 or are already interested in total synthesis. In any case, this brief commentary will explain the concept very simply. For advanced content, check out this page or my videos.
Imagine you’re a pastry chef and have stumbled across a picture of the most delicious-looking cake – but you lack the recipe. How can you re-create the culinary masterpiece? It will take some planning (which ingredients do I need?), experimentation (what baking temperature, for how long?), practical skills(how do I pipe the frosting?)…
… and luck! As we will see, some chemists had to push the ambiguity to the limit – sometimes testing a thousand reactions for one step, or realizing the shape of their glassware influenced their product!
Cake = Complex molecules
Chemists and the world are interested in complex molecules – a chemist’s cake – with intriguing properties – the cake’s flavours. For instance, natural products produced by other organisms can have biochemical activities that make them candidates for potential medicines.
What is total synthesis? Simply said, it is the complete chemical assembly of complex targets from simpler starting materials – like baking a cake!
Let’s take lissodendoric acid A as an example. This marine natural product isolated from sponges contains several connected rings and functional groups (in blue: amines, double bonds, carboxylic acid). This unique molecular setup gives the molecule anti-Parkinson’s disease activity in certain model experiments. Once chemists are sufficiently convinced of such a target‘s utility – or sometimes simply its molecular beauty – they need to figure out how to synthesize it. Note: In some cases, large-scale isolation from natural sources or biotechnological production might be feasible.
Recipe: Retrosynthetic analysis
Much like chefs writing recipes, chemists design a planen route to their masterpiece. This roadmap details the individual steps of reactions needed to transform simple starting materials into the desired molecular delicacy.
This plan is defined in a backward sense and thus called retrosynthetic analysis. This means that the first retrosynthetic “disconnection” corresponds to the finalstep of the forward/ laboratory synthesis.
Why backwards? Imagine you need to replicate a cake based on an image. Your first thought will be “How do I finish off the top and add the glazing?” (i.e., last ‘forward’ step). Later, you might think “How do I assemble the various layers of the cake?” (i.e., building the cake’s complexity).
You would not directly jump into thinking “For this cake, I need exactly 10 cups of flour and 8 eggs”. This would be a premature move! You haven’t even thought about how many cake layers you need, what their consistency and thickness should be…
How a retrosynthetic analysis looks like in practice. The target natural product (+)-Heilonine (1) is believed to be a constituent in the important Chinese herbal drug “Bei-mu”, which has traditionally been used as a sedative, antitussive, and expectorant. Source: JACS 2021, 143, 40, 16394|CC-by-4.0
It’s the same in total synthesis! We first work backwards through plausible, theoretical retrosynthetic disconnections. With this, we arrive at suggested starting materials for our laboratory endeavours in real life.
Experimentation: How To Learn Chemistry
Just like baking, chemistry is highly experimental. How can you tell if baking your cake layers at 170 °C, 180 °C, 190 °C or 200 °C will work best?
1) The first source is intuition to narrow your option space. For instance, baking the layers at 100 °C will either not work or take way too long. Baking them at 300 °C will lead to a bit too much crunch 😉 ! You don’t have to try 300 °C as the outcome is already certain.
Just like a freezing flame, thinking about the deprotonation of ketones with cyanide does not make sense. Once you have studied enough theory and reviewed practice, it becomes second nature!
As an example, we know that certain deprotonation reactions will simply not work if our base is not “strong”/basic enough. This is why pKa values are important! Chemists acquire their “common sense” by studying the fundamentals of chemical concepts, reactions and synthesis. Additional intuition is gathered by reading about and performing experiments. With time, chemists get a sense of what will likely work vs. what will not.
2) The second source is existing external research. Let’s assume other chefs reported 180 °C to work well for cakes with similar albeit different ingredients. This means starting at 180 °C is a good idea.
Let’s assume a specific Diels-Alder reaction was reported to require elevated temperature (e.g., reflux in toluene). If we want to perform a very similar reaction, we can assume that we don’t need to test cold temperatures like – 78° C. By reviewing scientific publications and patents, chemists can derive starting points for their experimentation.
3) The third source are your own experiments. If you want to bake a cake no one has ever baked before, you will need to gather completely novel data points.
Let’s pick up the example of (+)-Heilonine again. As reported by the authors, use of standard conditions (data source #2, if you will) was not fruitful. Instead, thorough investigations were need to identify adequate reaction conditions for this unique educt. Such optimization efforts also draw on intuition and inventiveness.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Is Chemistry Random?
Chemistry is only predictable to a certain (low) degree. Finding successful reaction conditions during organic synthesis can be challenging or straight-up impossible!
“Roughly 1000 experiments were conducted changing every conceivable variable from the base used to deprotonate, the solvent employed, additives, and the electrophile. Emerging from this exhaustive study was the remarkable finding that the addition of LaCl3·2LiCl to the extended sodium enolate of 3, followed by quenching with freshly prepared formaldehyde gas led to the desired adduct 11 in 84% yield as a 2:1 diastereomeric mixture favoring 11 (3 g scale).“
From: 11-Step Total Synthesis of (−)-Maoecrystal V | JACS 2016, 138, 30, 9425
Finding gold nuggets can sometimes be very tedious and serendipitous. Equally random are findings like the following:
Reproduced from Chem. Sci., 2022,13, 6181 with permission from the Royal Society of Chemistry.
What you are seeing is a reaction that only works in old borosilicate reaction flasks. This can happen in cases where the glass surface of a flask is actually where the reaction occurs.
Similar to such “vessel effects”, the source of chemicals (i.e., the supplier/ vendor) can in rare cases also make or break reactions. This can be driven by trace metal impurities which catalyse or also poison certain reactions. These impurities can vary across suppliers due to different syntheses/ purifications of these chemicals. Imagine if you could only bake your most beloved cake with a certain brand of milk!
Aside from driving chemists crazy, such unexpected effects exemplify the experimental nature of chemistry. Navigating this uncertainty while using strategic planning, creativity and practical skills – this makes total synthesis an art!