The Diels Alder reaction is a [4+2] cycloaddition reaction between a conjugated diene and a dienophile. Here we explain its mechanism, orbitaltheory, stereoselectivity and endo rule using 3D models and examples.
Table of contents
Mechanism: The Diels Alder reaction is a concerted [4+2] cycloaddition of a diene in its s-cis conformation with a dienophile.
Selectivity: Diels Alder reactions can be regioselective and stereoselective. As there are no intermediates, they are also stereospecific.
Substituent effects: Normally, a nucleophilic, electron-rich diene reacts with an electrophilic, electron-poor dienophile. Resonance explains regioselectivity, while secondary orbital overlap explains the endo rule.
Orbitals: In the normal electron demand DA reaction, the diene’s HOMO reacts with the dienophile’s LUMO. A smaller energy gap (due to substituents, Lewis acids) increases reactivity.
Hetero Diels Alder reactions: Involve reactants with non-carbon heteroatoms
Inverse electron demand Diels Alder: Feature electron-poor dienes and electron-rich dienophiles.
Remember: DA is often the easiest way to synthesize 6-membered rings!
1. reaction mechanism
As the Diels Alder is a pericyclic reaction, it proceeds in one step / concerted. The diene, containing two conjugated alkenes, reacts with a dienophile which is usually an alkene or alkyne (the name means something that likes dienes; sorry, chemistry is strange). Both reactants are flat (double bonds and diene is conjugated) and approach each other in a face-to-face manner.
The driving force of the Diels-Alder reaction is the formation of two σ-bonds (σ-bonds are stronger than π-bonds) in the cyclohexene product. The four atoms linked to these sigma bonds all change from sp2to a sp3 hybridization in the product.
Note that the cycloaddition initially gives the cyclohexene in its boat conformation. Try to really visualize how the transition state. If the dienophile approaches from the bottom (as drawn here; the alternative approach from the top is equally likely for achiral molecules), we form the boat conformation where the hydrogen that was previously pointing away from the diene, the Hexo, “looks up” (wedge in the product). As we see below, the reaction is stereospecific so both Hexo and Hendo in the diene retain their relative orientation (i.e., they look the same way in the product).
The diene has to be in the s-cis conformation to react (bonds form simultaneously).
Unlike π-bonds in alkenes, the single bond can rotate and thus change its conformation. However, given the steric interactions that the CH2 groups experience when they are on the same same of the bond, the s-cis conformation is higher energy than s-trans. The energy difference is ~2.9 kcal/mol [ref 1], so only ~1-2% of butadiene molecules are actually in the s-cis form at a given time.
Note that some dienes like cyclopentadiene are extremely reactive because they are locked in the s-cis conformation.
By the way, we do not say [4+2] because 4 electrons from the diene react with 2 electrons from the dienophile. This is indeed the case – the transition state can actually be considered to be aromatic – but [4+2] refers to the number of atoms involved. For instance, there are so-called [3+2] dipolar cycloadditions which also operate with 6 electrons in total, but only 5 atoms are involved.
Chemistry would be so easy without substituents! By adding substituents on the diene and dienophile, we start to observe reaction selectivity and increased or decreased reactivity. Let’s first understand this in theory, and then check out the specific substituent effects.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
2. Selectivity in Diels Alder reactions
Diels Alder reactions are usually regioselective, stereospecificand stereoselective.
Depending on which atoms of the diene and dienophile make contact, we can get two regioisomers (A is either next to E, or A is next to F). The actual regioselectivity is driven by the nature of the substituents (see below).
Because 1) there are no intermediates in the Diels Alder, and 2) both σ-bonds are created from the same side/face, the relative orientation of substituents is fully retained in the product. This is why the reaction is stereospecific.
Beyond the different orientation leading to regioselectivity, the dienophile can also be oriented outside or inside of the diene. This again gives two different products, exo and endo. Note that the example here shows just one regioisomer (it does not show products where A is next to F).
3. Substituent effects in diels alder reactions
Substituents critically influence selectivity and reactivity of Diels Alder reactions.
Normal electron demand DA reactions are cases where the diene is electron-rich, while the dienophile is electron-poor. Typically, the diene has electron-donating groups (EDG; e.g., -OMe) while the dienophile has electron-withdrawing groups ( EWG; e.g., -CO2Me). The majority of reactions follow this polarity, so let’s focus on such examples first. The opposite happens in inverse electron demand DA reactions (explained further below).
The regioselectivity of Diels-Alder reactions can be predicted by considering potential resonance structures of the two reactants. ➡️ The most nucleophilic position of the diene will prefer to add to the most electrophilic position of the dienophile.
Diels Alder reactions can give endo (kinetically preferred) or exo products (thermodynamically more stable). ➡️ If the dienophile carries substituents with π-electrons, the endo product is usually preferred. (explanation requires orbital theory, see below) ➡️ If you are struggling with drawing endo versus exo, draw the transition state in 3D (e.g., as here, diene adding from the top) and draw the resulting boat conformation to compare which groups point which way.
4. Frontier Molecular Orbital (FMO) Analysis
The Diels Alder reaction, just like many other reactions, can be explained by purely looking at the so-called frontier molecular orbitals (FMO). These are the HOMO (highest occupied molecular orbital; filled) and LUMO (lowest unoccupied molecular orbital; empty) of the π-system.
The Diels Alder reaction is exergonic (ΔG<0) – the Hammond postulate tells us that such a reaction has a relatively “early” transition state that looks similar to the starting materials. Thus, the orbitals of the starting materials are a good “proxy” for what will happen in the transition state.
In case you cannot construct the MO diagram for butadiene or ethylene, I refer you to other web sources that explain this.
In the normal electron demand scenario, the HOMO of the diene reacts with the LUMO of the dienophile. Notice how the orbital lobes (positive and negative, grey and black) that overlap have the same phase.
The closer the orbitals in energy (small HOMO-LUMO energy gap), the easier the reaction! This is because when orbitals of similar energies overlap, they experience a higher stabilization of the newly formed orbitals. We want as high of a stabilization as possible, as this decreases the energy of the electrons that we use to populate the new molecular orbital.
Electron-donating groups raise the energy of all molecular orbitals; electron-withdrawing groups decrease the energy of all molecular orbitals.
By having an electron-rich diene and an electron-deficient dienophile, we lower the HOMO-LUMO energy gap (new green line). This means the stabilization experienced by orbital interaction for the purple electrons will increase (they will go down in energy, which is good). This effect is additive – the more substituents we add, the easier and faster the reaction.
The stereoselectivity and endo rule can also be explained through orbital considerations. If the dienophile is oriented endo, the π-orbitals of the carbonyl group (or any other group that has π-system) can interact with the “central” diene orbitals. This lowers the transition state (TS) of the endo approach compared to the exo approach – and thus, the two substituents in our example above point in the same direction.
This might be a bit clearer from the 3D model here. Ignore the non-planarity of the butadiene (my orbital computation was trolling me).
Substituents do not only influence the energy of molecular orbitals, they also change the orbital coefficients at the different atoms. A larger coefficient means that an orbital is relatively more localized on a given atom – e.g., for the HOMO, the atom with the largest coefficient is most nucleophilic. This explains the regioselectivity of Diels Alder reactions more truthfully than the resonance structures which we have mentioned above. However, because this article is not intended to be fully exhaustive, we will not explain coefficients in detail here.
For dienophiles with basic sites on electron-withdrawing groups, Lewis acids can function as catalysts and increase the regio- and stereoselectivity. Coordination of the Lewis acid makes the dienophile even more electron-deficient, and increases the relative polarization (and orbital coefficient).
5. Hetero Diels Alder reaction
DA reactions are not limited to only carbon atoms. Both the diene or the dienophile can feature non-carbon heteroatoms. An example for such hetero Diels Alder reactions is the cycloaddition above with an aldehyde as the dienophile. This works well with the normal electron demand scenario as the carbonyl π-system has a low-energy LUMO.
6. Inverse electron demand Diels Alder
Inverse electron demand Diels Alder reactions are cases where the diene is electron-poor, while the dienophile is electron-rich. Now, the HOMO and LUMO are mapped the other way around: the electron-rich dienophile is nucleophilic and has a high-energy HOMO – while the electron-poor diene is electrophilic and has a low-energy LUMO.
Typically, inverse electron demand reactions as part of multi-step cascade reactions.
Draw the correct product for the given Diels Alder reaction!
So the heteroaromatic ring clearly has a diene, but where is our dienophile [ref 2]? The first step of the reaction is a condensation-enamine formation of the amine with our ketone. The enamine is an electron-rich olefin, so a perfect dienophile in the inverse electron demand scenario. To figure out which regioisomer will be preferred, we need to consider which resonance structures are stabilized by the EDG and EWG, respectively.
After the first Diels Alder reaction, we have a twist: The N=N group can be kicked out in a reverse or retro Diels Alder reaction. It works the exact other way around, with the very stable N2 driving the reaction forward (irreversible step). The last step is aromatization to the much more stable product after elimination of pyrrolidine. I will work on a separate article on the retro Diels Alder reaction in future.
If you learned something, feel free to check out my page or my videos!
To a Schlenk tube charged with 3,5-dibromo-2-pyrone [diene] (20, 561 mg, 2.21 mmol) in 7 mL of toluene was added alkenyl boronate [dienophile] (600 mg, 1.70 mmol) at rt. The resulting mixture was heated in an oil bath at 110 °C for 3 days. The reaction mixture was cooled to rt, concentrated in vacuo, and chromatographed (20:1 hexane/EtOAc → 5:1 hexane/EtOAc) to give cycloadduct as a white solid (720 mg, 70%).
Diels Alder reaction references
[1] Thermodynamics of conformational change in 1,3-butadiene studied by high-temperature ultraviolet absorption spectroscopy | Philip W. Mui, Ernest Grunwald | J. Am. Chem. Soc. 1982, 104, 24, 6562–6566
[2] Inverse electron demand Diels-Alder reactions of heterocyclic azadienes: formal total synthesis of streptonigrin | Dale L. Boger, James S. Panek | J. Am. Chem. Soc. 1985, 107, 20, 5745–5754
[3] Total Synthesis of (±)-Clivonine via Diels–Alder Reactions of 3,5-Dibromo-2-pyrone | Cheng-dong Wang, Qinyang Chen, Seunghoon Shin, Cheon-Gyu Cho | J. Org. Chem. 2020, 85, 15, 10035–10049
👀 Left: 3D model of the simplest Wittig reagent (methylenetriphenylphosphorane)
Wittig reaction mechanism
Step 1 – Wittig reagent generation: Every Wittig reaction is based on a carbonyl compound and a Wittig reagent. This is a phosphonium ylide species that can be drawn in two resonance structures: the neutral phosphorane structure or the ylide structure. Chemists generate this species by deprotonating a precursor, a phosphonium (positive charged phosphorous) salt, with strong base. The choice and strength of the base depends on the stabilization of the Wittig reagent (see below). A Wittig reagent that can stabilize the negative charge through other groups can be formed by using a milder base.
How does this work in the laboratory? To avoid cross-reactions (you might know that carbonyls can also be deprotonated by strong bases), chemists add the carbonyl to the reaction only after the base was used to deprotonate the phosphonium. At the very bottom, you can find two exemplary procedures. Some Wittig reagents are so stable that they can be isolated.
Step 2 – Olefination: The ylide/phosphorane is very nucleophilic at the carbon, so it can intermolecularly attack the electrophilic carbonyl carbon. At the same time, the oxygen is nucleophilic and attacks the electrophilic phosphorous. This step likely occurs in a one-step [2+2] addition[see reference 1]. The four-membered ring product is called an oxaphosphetane.
The ring can fragment the ‘opposite way that it was made’ – releasing triphenylphosphine oxide (the driving force) and our alkene product. This is why the Wittig reaction can be classified as an olefination reaction.
The orientation of substituents at the oxaphosphetane determines the stereoselectivity of the reaction. There are two chiral centres on the ring – so cis and trans diastereomers are formed. The stereochemistry of the oxaphosphetane translates into the product following the retro-[2+2]. Cis leads to (Z), trans leads to (E). Here, we note that because the Wittig reagent is a so-called non-stabilized ylide, the (Z) olefin is preferred (see below).
What type of betaine is this? What diastereomer do you expect in the product?
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
What is the Driving force in the Wittig Reaction?
This is a common question for students. The driving force of the Wittig reaction is the oxidation of triphenylphosphine to form triphenylphosphine oxide or Ph₃P=O. This new phosphorus-oxygen double bond is very strong, making its formation highly favorable from a energetic (thermodynamic) standpoint. You could say there is also a kinetic driving force as the ylide reagent is highly nucleophilic and thus, reactive with the electrophilic carbonyl starting material.
Stabilized ylides and non-stabilized ylides
Depending on the substituents attached to the α-carbon (next to the phosphorous), ylides are categorized into stabilized or non-stabilized ylides. The stability refers to the ability of the substituent to stabilize the negative charge.
Stabilized ylides have electron-withdrawing groups (EWGs) attached to the α-carbon. These can include carbonyls like esters, or nitriles. The mesomeric effect of electron-withdrawing groups stabilizes the negative charge through resonance. This makes the Wittig reagent less reactive but more selective, usually favouring the more thermodynamically stable (E)-alkene product. These Wittig reactions can operate at higher temperatures.
Non-stabilized ylides lack such electron-withdrawing groups and feature alkyl groups. Due to the lack of stability (there is no resonance with alkyl groups), non-stabilized ylides are more reactive. They usually form the kinetically favoured (Z)-alkene product. To maintain stability and the kinetic selectivity, reactions with these ylides are performed at low temperatures.
Semi-stabilized ylides aryl or alkenyl substituents. They fall between the stabilized and non-stabilized ylides and their stereoselectivity is typically poor, leading to similar (E) and (Z) alkene mixtures.
Question for you: Which type of ylide is easier to form by deprotonation? What are their rough pKa values in DMSO?
➡️ Stabilized ylides are more easily formed by deprotonation as they are by definition compounds that stabilize the negative charge on carbon. This means the respective conjugative acid has a lower pKa value (i.e., is more acidic).
Is the Wittig Reaction Concerted or Step-wise?
Over much of its history, the Wittig reaction has been described as a stepwise ionic process. Instead of concerted (one-step) [2+2] cycloaddition, it was assumed that the addition to the carbonyl proceeds step-wise, forming a betaine intermediate. However, modern research suggests that the cycloaddition is more likely [ref 1].
But hey, if your course teaches you the step-wise one, just write that one. Rather get full marks than trying to be right and a smart ass 🙂
Wittig Reaction: Advanced Example [Ref 2]
Here’s a final question (2nd year undergrad level):
What is the mechanism of this two-step sequence? Is the ylide stabilized?
The answer is given below to avoid spoilers.
If you liked this post, feel free to check out other articles on my page or my educational videos!
Under an N2 atmosphere, methyltriphenylphosphonium bromide (40 mg, 0.113 mmol) was suspended in dry THF (1 mL) in a Schlenk tube at 0 °C. BuLi (2.5 M in hexane, 45 μL, 0.113 mmol) was added dropwise. After stirring for 30 min at this temperature, the mixture was cooled to –78 °C, compound 20 (22 mg, 0.057 mmol) was added dropwise as a solution in THF (1 mL). The reaction was continued at the same temperature for 1 h and then at 0 °C for 30 min. Water (1 mL) was added to quench the reaction and the mixture was extracted with CH2Cl2 (3 × 2 mL). The organic extracts were then combined, dried with anhydrous Na2SO4, filtered, concentrated, and purified with by chromatography on silica gel (pentane/EtOAc, 2:1) to give compound 21 (22 mg, >99 % yield).
To a solution of aldehyde (+)-17 (1.40 g, 2.81 mmol, 1 equiv) in dry THF (35 mL) was added crystalline (α-carbomethoxyethylidene)triphenylphosphorane (1.96 g, 5.62 mmol, 2 equiv). The reaction mixture was stirred and heated from room temperature to 50 °C for 2 h under argon. The solvent was then removed under reduced pressure. The residue was purified by flash chromatography (PE/EtOAc 4/1) to give the ester (+)-18 as a white solid (1.31 g, 82%).
Wittig Reaction answer [Ref 2]:
The advanced example above is a Wittig reaction, followed by a Claisen rearrangement. The latter is a [3,3]-sigmatropic rearrangement, a type of pericyclic reaction. It converts allyl vinyl ethers to γ,δ-unsaturated carbonyls. This is an interesting transformation as it looks like it preserves the aldehyde, but moves it one carbon further away from the aromatic ring.
The ylide substituent (-OR) is a mixture of electron-withdrawing (sigma-effect) and electron-donating (mesomeric effect). Thus, one can argue the ylide is semi-stabilized. This is reflected in the rather close ratio of (E) to (Z) alkene after the Wittig. Fortunately, the mixture is not an issue as the Claisen rearrangement gives the same product regardless of diastereomer in this system.
[3] Concise Total Synthesis of Dihydrocorynanthenol, Protoemetinol, Protoemetine, 3-epi-Protoemetinol and Emetine | Shuangzheng Lin, Luca Deiana, Abrehet Tseggai, Armando Córdova | European Journal of Chemistry 2012, 2012, 2, 398-408
[4] Total Synthesis of (−)-Chanoclavine I and an Oxygen-Substituted Ergoline Derivative | Jia-Tian Lu, Zi-Fa Shi, Xiao-Ping Cao | J. Org. Chem. 2017, 82, 15, 7774–7782
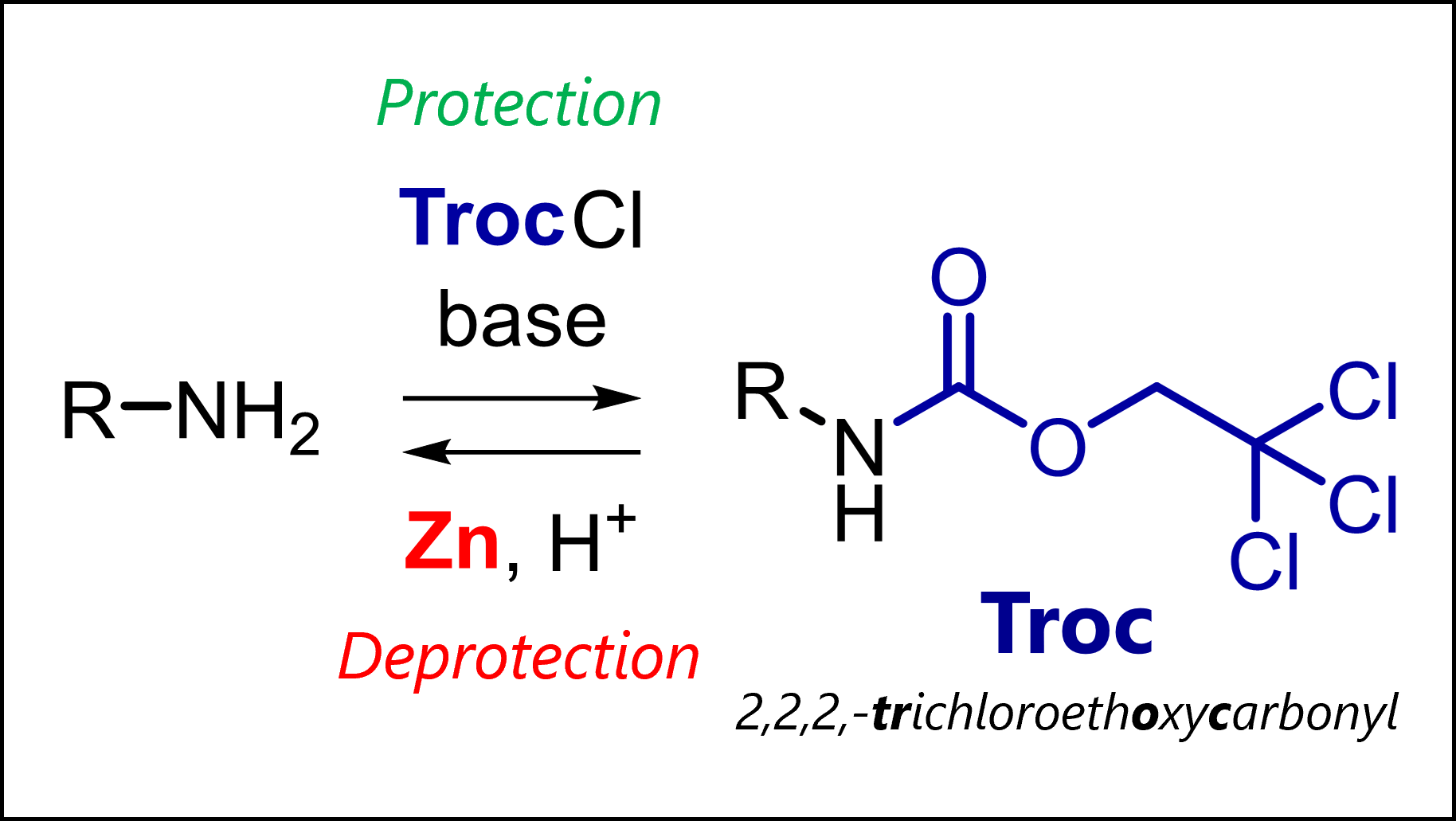
The Troc protecting group protects amines in organic synthesis. Troc is introduced with TrocCl and deprotected by reduction (Zn).
🫡 Hello! Here’s what you’ll learn here:
Table of contents
👀 Left: 3D model of Troc (chlorines in green!)
What is the Troc Protecting Group?
Troc (2,2,2-trichloroethoxycarbonyl) can convert amines or alcohols into stable carbamate or carbonate derivatives. This leads to a similar effect that we see in groups like Fmoc or Boc: the previously nucleophilic amine or alcohols loses its reactivity (driven by delocalization of electrons into the carbonyl system).
The nice thing is that Troc is orthogonal as the deprotection conditions for Fmoc (base), Boc (acid) or silyl groups like TBS (fluoride) do not remove it. Instead, it has a unique mechanism of reductive beta-elimination to release the free group.
Did you know: The Troc group was introduced by the legendary Robert Burns Woodward in the 1960s? (see below)
Troc Protection Mechanism
The Troc protecting group is introduced by reacting the free amine or alcohol with 2,2,2-trichloroethyl chloroformate (Troc-Cl) with addition of a base.
Most common protection conditions are Troc-Cl with pyridine in CH2Cl2 or THF. If the starting material is very polar, Troc-Cl with NaOH or NaHCO3in water.
Troc Deprotection Mechanism
The Troc protecting group can be removed with certain reductive methods which all function via beta-elimination. Most common is use of zinc or other single-electron reductants that reduce the terminal carbon. The beta-elimination gives a free carbamate – an intermediate seen in mechanisms of many other protecting groups – that rapidly decarboxylates to the deprotected amine (or alcohol).
Most common deprotection conditions are Zn powder in THF/H2O or so-called couples/alloys consisting of mixtures of Zn-Pb or Cd-Pb. More rarely used, reduction with electrolysis also removes the Troc group.
Always remember protecting group stability or lability are always general (e.g., here: removal by reduction). Nothing in chemistry (or life) is black and white 🙂 Here is a neutral method (not reductive) using trimethyltin hydroxide. [1]
A fascinating point is that under the right conditions, Me3SnOH actually deprotects Troc selectively in the presence of methyl esters (you would expect that the methyl ester is quite labile to hydrolysis with hydroxide).
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Troc protecting group in total synthesis
The very first introduction of the Troc protecting group was already… very advanced! Woodward and co-workers used the starting material below in the synthesis of cephalosporin C, an antibiotic natural product. [2]
What is the product after treating this molecule with Zn in aqueous AcOH?
This is a cool example where just one of the three groups decarboxylates. The others are not true Troc groups but were instead masking the free acid. If we do not have a free carbamate or carbonate after reductive elimination, we do not see a decarboxylation.
Below you can find typical Troc protection and deprotection conditions.
Troc Protection conditions [3]
To a solution of the alcohol (0.80 g, 1.48 mmol) in methylene chloride (30 ml) at 0 °C was added pyridine (0.96 ml, 11.84 mmol, 8 equiv) followed by 2,2,2-trichloroethyl chloroformate (0.8 ml, 5.92 mmol, 4 equiv), and the reaction mixture was stirred at 0 °C for 1 h. Saturated aqueous sodium bicarbonate (50 ml) was added and the organic layer was separated. The aqueous layer was extracted with methylene chloride (3 x 50 ml), and the combined organic layers were dried over anhydrous sodium sulfate and concentrated in vacuo. Purification by flash column chromatography (2% EtOAc/hexanes) afforded protected product (0.98 g, 93%) as a colorless oil.
Troc deprotection conditions [4]
To a solution of protected Troc-amine (40 mg, 57 µmol) in 4 mL of MeOH was added activated zinc (400 mg). The mixture was stirred at 25 °C for 5 min, and glacial HOAc (4 mL) was added. The mixture was heated at 60 °C for 30 min, cooled and concentrated under reduced pressure. The residue was treated with 5 mL of 5% aqueous NaOH, and the solution was extracted with EtOAc (5 × 5 mL). The combined extracts were washed with brine, dried over anhydrous K2CO3, and concentrated under reduced pressure. Flash chromatography on silica gel (100:1 CH2Cl2/MeOH) gave 25 mg (86%) of the free amine as a viscous oil.
TROC Protecting Group References
P. G. M. Wuts, T. W. Greene: Greene’s Protective in Organic Synthesis (Wiley)
[1] Highly Chemoselective Deprotection of the 2,2,2 Trichloroethoxycarbonyl (Troc) Protecting Group | Barry M. Trost, Christopher A. Kalnmals, Jacob S. Tracy, and Wen-Ju Bai | Org. Lett. 2018, 20, 8043−8046
[2] The Total Synthesis of Cephalosporin C. | Woodward, R. B.; Heusler, K.; Gosteli, J.; Naegeli, P.; Oppolzer, W.; Ramage, R.; Ranganathan, S.; Vorbruggen, H. | J. Am. Chem. Soc. 1966, 88, 852− 853
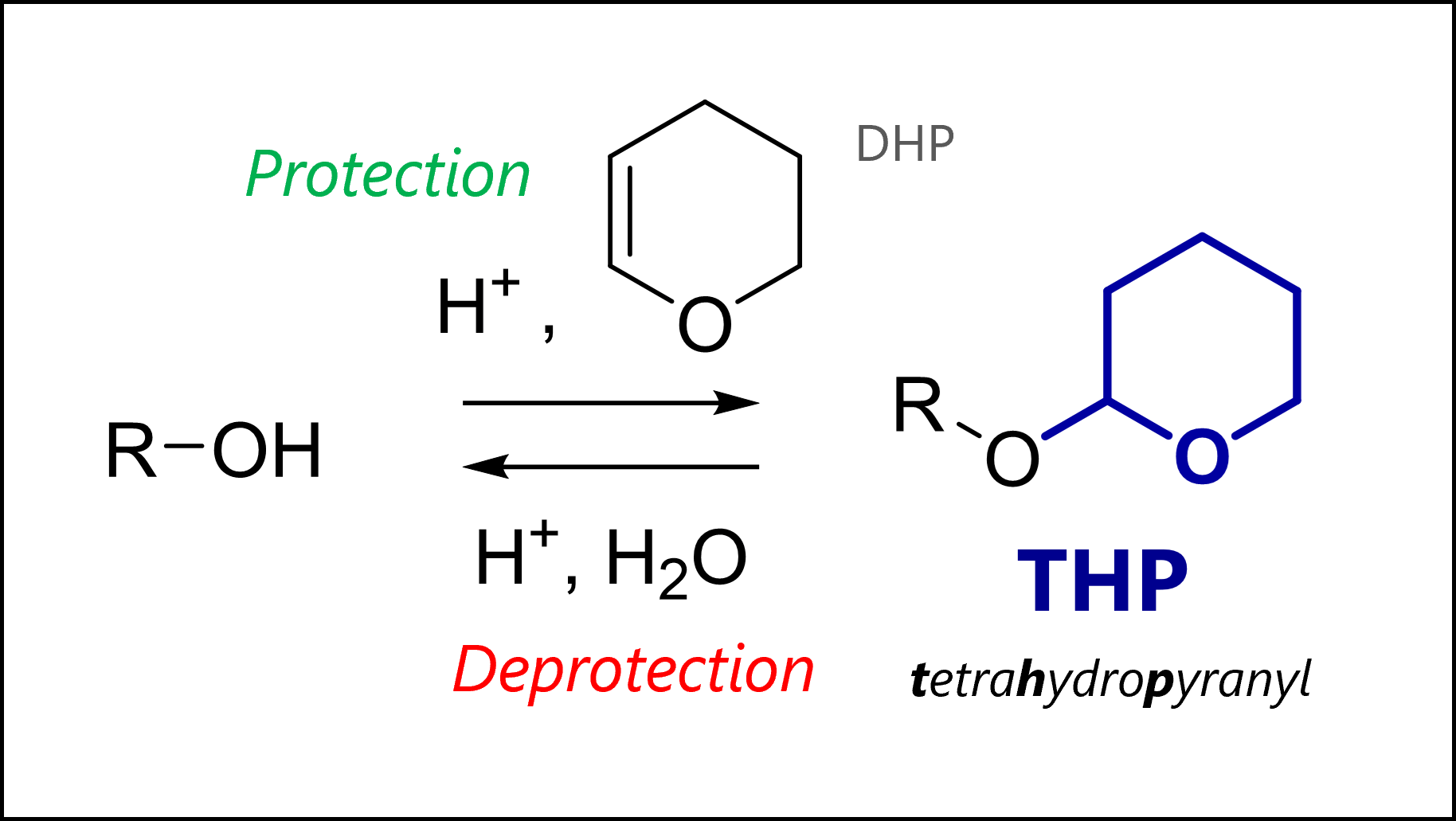
Tetrahydropyranyl ethers were one of the first protecting groups for alcohols. Nowadays, they are seen less commonly, though still used. The THP group is easily removed under acidic conditions (mechanism below) and stable to organometallic nucleophiles, electrophiles (as the protected oxygen is less nucleophilic), reduction or base. The protected THP ethers are actually a type of acetal (‘double-ether’).
THP Protection Mechanism
THP protection uses acid catalysis and 3,4-dihydro-2H-pyran or DHP. The mechanism proceeds by THP pre-activation with acid, leading to a stabilized cation. Here, the oxonium is drawn but you can imagine the other resonance form with the positive charge on the carbon which is ultimately where the ion is most electrophilic. Our free hydroxyl group then attacks the carbon in a nucleophilic addition, and loses a proton to give the protected THP ether. The last step regenerates our acid catalyst.
The most common protection conditions are catalytic TsOH or pyridinium p-toluenesulfonate (PPTS, a form of TsOH with lower acidity) together with 3,4-dihydro-2H-pyran in dichloromethane.
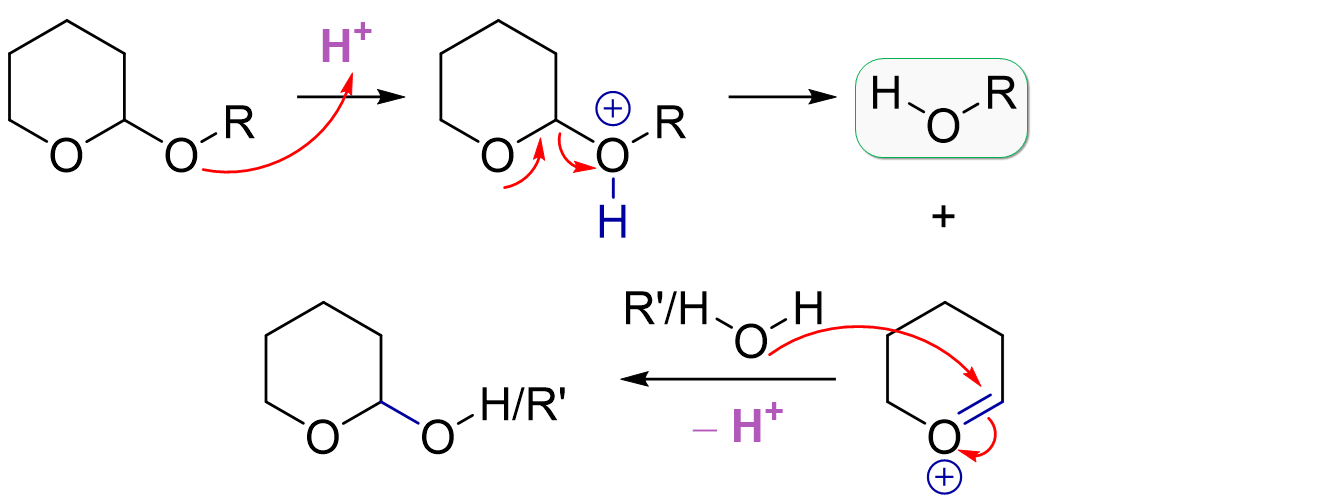
THP Deprotection Mechanism
THP deprotection is similar to MOM deprotection and actually also similar to THP protection. Acid catalysis activates the acetal system towards dissociation of our initially protonated alcohol. Again, it’s the same stabilized cation intermediate but based on the choice of solvent used, we have different potential byproducts. The solvent is obviously present in large excess, so it will preferentially attack the carbocation instead of our just liberated hydroxyl group. For example, methanol gives the methyl-substituted THP ether while use of water would give the free hydroxyl group (this can open to the linear aldehyde).
The most common deprotection conditions are AcOH:THF:H2O or PPTS in EtOH.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
THP protecting group diastereomers
One of the drawbacks of the THP protecting group versus the TBS protecting group, beyond its lower stability, is that it introduces a second chiral center. If our starting material has already at least one chiral carbon, we form diastereomers. This can complicate the separation and identification (e.g., NMR) of products – because as you know, diastereomers have different physicochemical properties.
Interestingly, some older research [1] tried to make use of this ‘drawback’. In this work, the chemists used a THP-derivative as a chiral auxiliary for nucleophilic additions to an aldehyde in the molecule.
In these derivatives, one side of the aldehyde is shielded from nucleophilic attack while the other is exposed. This leads to very high diastereoselectivity at the newly formed carbon (a tertiary alcohol). It’s not terrible useful but interesting that a protecting group can be used to exert diastereoselectivity. You could imagine this potentially being useful in some complicated total syntheses.
“To a 100-mL, single-necked, round-bottomed flask equipped with a magnetic stir-bar, argon inlet with septum, was charged with dihydropyran (1.5 equiv), followed by CH2Cl2 (15 mL) and PPTS (177.4 mg, 0.706 mmol, 0.1 equiv). The contents were cooled to 0 °C in an ice-bath. Then a suspension of iodobenzyl alcohol 9 (2.08 g, 7.06 mmol) in CH2Cl2 (10 mL) was added at 0 °C over 10 min. After addition, the contents were warmed to rt. Dihydropyran (0.97 mL, 10.59 mmol, 1.5 equiv) was added again to the mixture after 30 min, because the starting material was still observed by TLC analysis. After another 30 min of stirring, H2O (50 mL) was added and the mixture was extracted with CH2Cl2 (3 x 50 mL). The organic layers were combined and washed with brine (2 x 50 mL), dried with Na2SO4, filtered and the solvent was removed under reduced pressure by rotary evaporation. The crude material was further purified using column chromatography (SiO2, 70 g; hexanes/EtOAc, 3:1) to afford (2.67 g, >99%) THP ether 26 as a colorless wax.”
THP deprotection experimental procedure [3]
“To a solution of alkene 12 (38.6 mg, 0.047 mmol) in 2-propanol (0.95 mL), p-toluenesulfonic acid monohydrate (21.7 mg, 0.114 mmol) was added at 0 °C and stirred for 17 h at room temperature. The reaction mixture was diluted with water, extracted with dichloromethane, washed with brine, and dried over sodium sulfate. The residue was purified by thin layer chromatography (hexane/ethyl acetate = 5/1). Alcohol 13 (34.6 mg, quant.) was obtained as a colorless oil.”
THP Protecting Group References
P. G. M. Wuts, T. W. Greene: Greene’s Protective in Organic Synthesis (Wiley)
[1] The tetrahydropyranyl group as a chiral auxiliary for the nucleophilic addition to α-alkoxy ketones | André B. Charette , Abdel F. Benslimane , Christophe Mellon | Tetrahedron Letters 1995, 36, 8557
[2] Total synthesis of (+)-papulacandin D | Scott E. Denmark, Tetsuya Kobayashi, Christopher S. Regens | Tetrahedron 2010, 66, 4745
[3] Total Synthesis of Eutyscoparol A and Violaceoid C | Takatsugu Murata, Takuto Iwayama, Teppei Kuboki, Shotaro Taguchi, Shou Tsugawa, Takumi Yoshida, Hisazumi Tsutsui, Ayana Shimauchi, Yukiho Kosaka, Isamu Shiina | Asian Journal of Organic Chemistry 2024, 13, e202400148
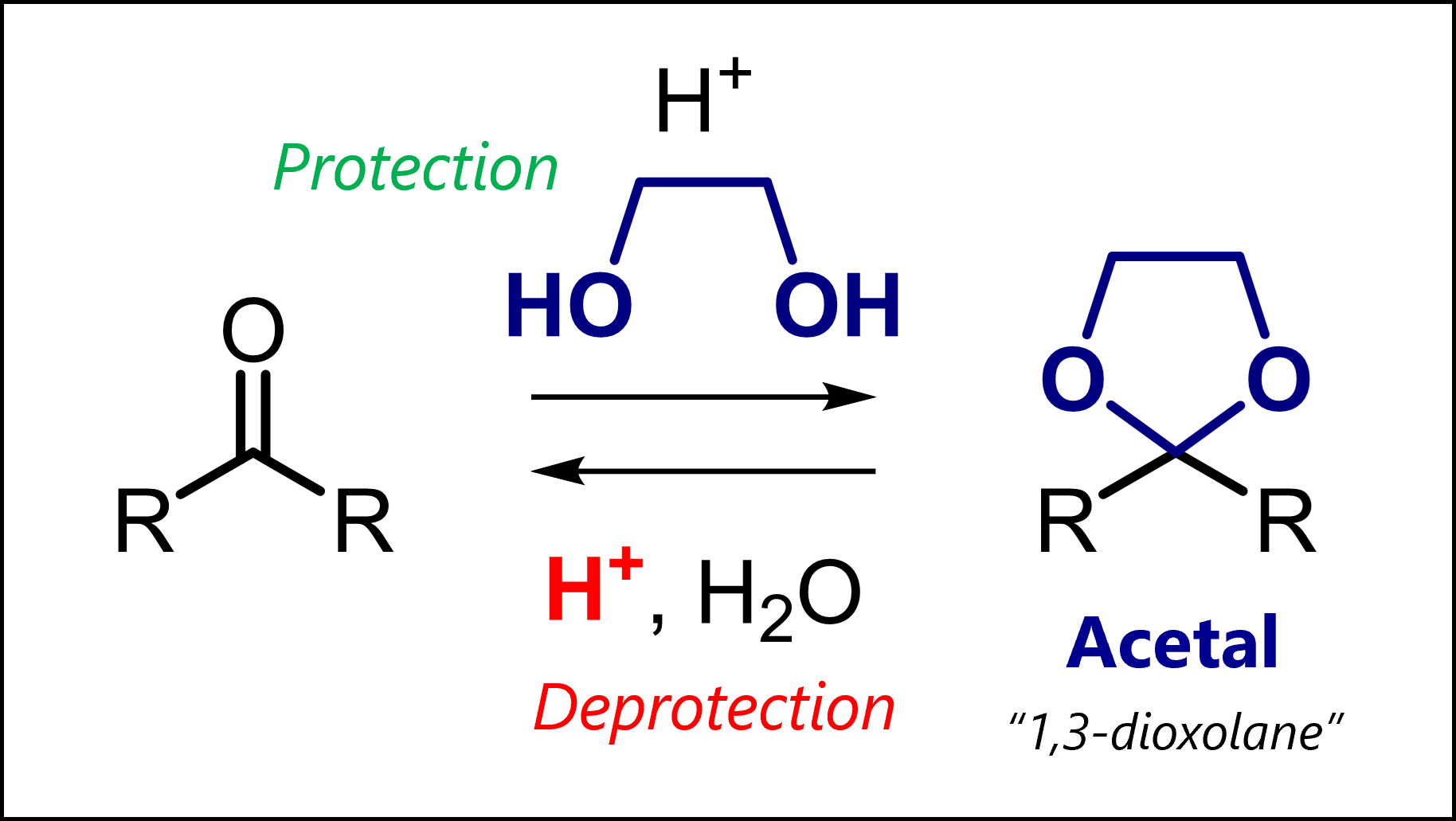
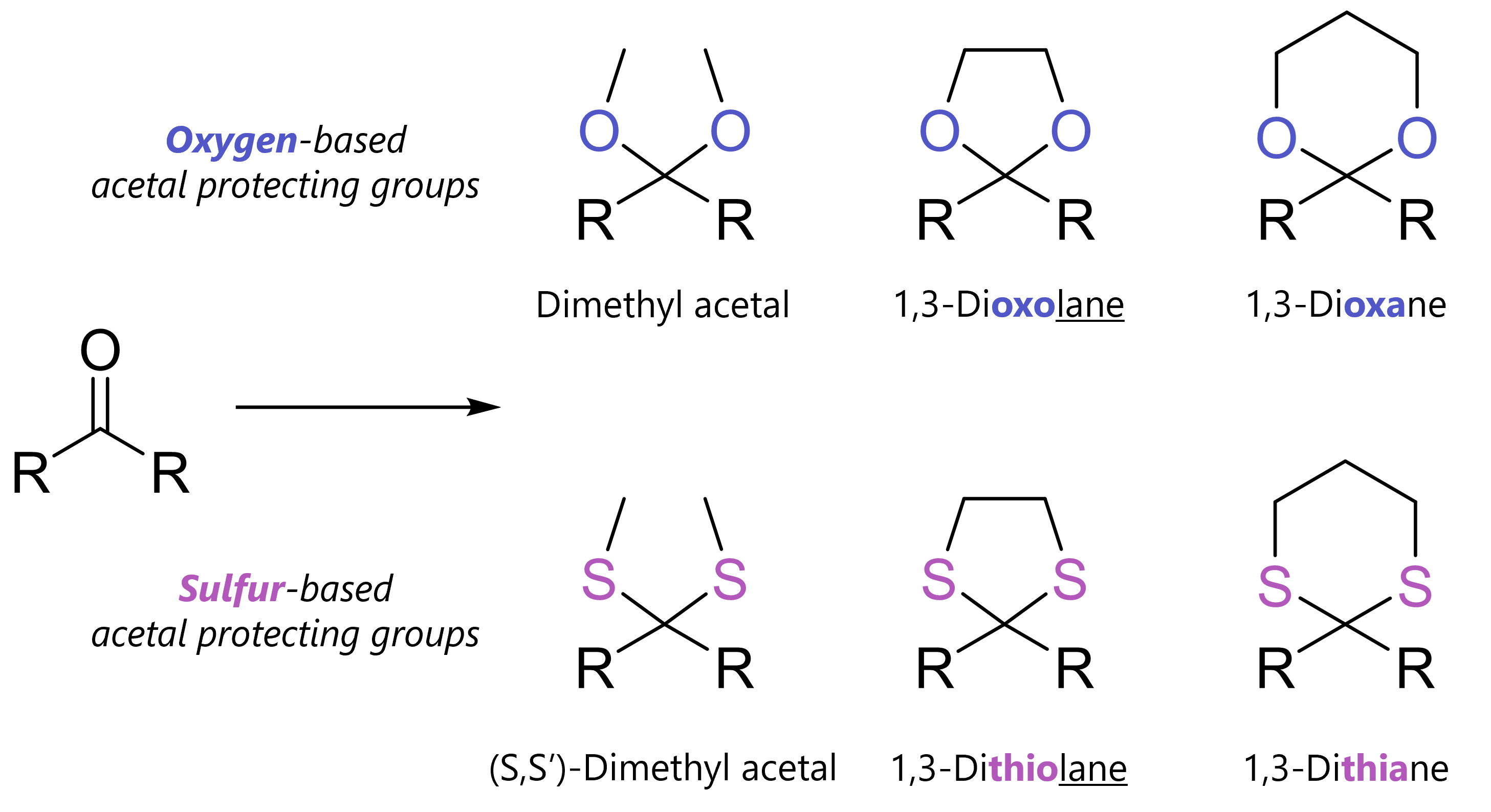
Acetal protecting groups protect carbonyls from bases, nucleophiles and hydride reduction. Among many variants, most common are dimethyl acetals, 1,3-dioxolanes and 1,3-dioxanes.
Table of contents
There is no single acetal protecting group! Rather, this is a broader family of similar protecting groups.
➡️ This article explains properties and mechanisms of acetals (protection and deprotection).
What Are Acetal Protecting Groups?
Acetals always protect carbonyl compounds. But how? This is where the variety can come from. On one hand, acyclic acetals form by reaction with an alcohol (-OH) or thiol (-SH) and catalytic acid. On the other, cyclic acetals form when carbonyls react with a diol or dithiol and catalytic acid.
Do you already know if cyclic or acyclic acetals are more stable? Why? (see below)
You will know that carbonyls are nucleophilic at the carbon. Any acetal protecting group renders it stable to these nucleophiles: aqueous and non-aqueous bases, organometallic reagents and hydrides. As always, we want to avoid unwanted reactions of one group (here, the carbonyl) to instead perform chemistry at another functional group. As we will see below, formation of acetals involves a two-step mechanism, including nucleophilic attack and subsequent dehydration, which drives the equilibrium towards product formation.
Difference between Acetal and Ketal
You might not be aware, but back in the day, people used to separate acetals – made from aldehydes – and ketals – made from ketones. Nowadays, acetal is the umbrella term that describes both – while ketal remains restricted to ketones (link to IUPAC definition).
Types Of Acetal Protecting groups
As mentioned, there are several relatives in the acetal protecting group family. The good thing is that they work very similarly!
=> You should simply know that acetals can be oxygen-based or less commonly, sulfur-based. The simplest acyclic acetal is the dimethyl acetal. Cyclic acetals have five-membered rings (1,3-dixolane; 1,3-dithiolane) six-membered rings (1,3-dioxane, 1,3-dithiane).
Acetal protection mechanism
As an example, 1,3-dioxolanes are prepared by treating carbonyls with ethylene glycol and acid. => Acetal protection or acetalization requires catalytic acid to activate the carbonyl (but only catalytic because the proton is regenerated in the final step) => Acetalization is a condensation as the original oxygen is kicked out as water
Typical conditions: Ethylene glycol and cat. TsOH (acid) in C6H6 as solvent at reflux.
Because every reaction is an equilibrium (imagine the arrows also going from right to left), chemists use ways to remove water from the reaction to ensure it can’t react back. For acetalizations, this involves using a Dean-Stark trap. The Dean-Stark trap is a glassware that collects water formed in a reaction through an azeotropic distillation. You might have heard about it – if not, does not matter. This is a physical removal – alternatively, dehydrating agents like trimethyl orthoformate can chemically remove the water by reacting with it (“scavenger”).
The same mechanism applies if we use other diols (e.g., to form six-membered 1,3-dioxanes), alcohols (e.g., methanol to form dimethyl acetals) or thiols to form sulfur-based acetal protecting groups.
Acetal Deprotection Mechanism
Deprotecting acetals is very similar to introducing them! The most common is an acid-catalyzed hydrolysis. Again, make sure you understand why it only requires catalytic acid (i.e., less than 1 “equivalent” of moles).
Typical conditions: Cat. pyridinium tosylate PPTS or HCl (as the acid) in a mixture of water (for the deprotection) and an organic solvent (to dissolve the starting material).
The sulfur-based acetals are special as they can also be removed with heavy metal salts – so Lewis acids like mercury(II) or silver(I) – or oxidants. The oxygen-based acetals are stable to these conditions. We will go into 1,3-dithianes and 1,3-dithiolanes into more detail in another post.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Acetal protecting group stability
Are cyclic or acyclic acetals more stable?
Cyclic acetals are more stable than acyclic ones. Why? Acidic hydrolysis starts with protonation (catalytic acid), and goes via the oxonium intermediate.
For the cyclic acetal, the newly released hydroxyl group is still in the same molecule – so the reverse reaction would be an intramolecular reaction which is very fast (entropically favored).
For acyclic acetals, formation of the oxonium cleaves off an alcohol as a separate molecule. Because the deprotection is in aqueous solvent, we have a lot of water molecules around. It is now much more likely water will attack the oxonium (leading to deprotection of the carbonyl) instead of the alcohol attacking. This is because we only have 1 molecule of alcohol formed for 1 molecule of starting material; on the other hand, we have a large excess of water molecules.
“Why do they call it organic chemistry? Nothing about this feels natural.” A student, somewhere out there
Organic chemistry is often perceived as one of the most challenging subjects for students. Unfortunately, this creates a lot of anxiety and misconceptions about its complexity and requirements, particularly for students in adjacent fields like medicine, pharmacy or biology.
But why the bad reputation?
Concepts: Yes, organic chemistry is complex! After all, it deals with the literal wizardry of transforming living matter. The complexity (and bad teaching) can make many students feel overwhelmed – amplified by cumulative learning: missing foundational concepts makes it difficult to understand advanced ones.
Representation: Organic chemistry requires abstract thinking and relies on molecular drawings that are often not intuitive or tangible. After all, we are drawing 3D things on 2D paper. Fischer projections anyone?! (I hated these) (how to change: look at 3D model, simulate it online, practice practice)
Process:Teachers rush through content without explaining it properly, leading students to fall behind completely and/or rely on memorization to stay afloat (how to change: try to maximize the support at your uni with open doors, ask more senior students – if no support environment, look online and try to understand the why, write as many things out as you can to build your mental muscle…)
But do not despair – organic chemistry can be more accessible than you might think. Here are some ways to help you address these issues.
These tips are not some crazy 200 IQ approaches, but they require putting in the work – thoughtfully. It’s like getting fit: it’s relatively obvious what you need to do (eat healthy, work out, sleep…), but the challenging part is doing it!
1. Organic Chemistry Concepts
Build the foundation: Whatever level of class you are taking, first go back to your older materials. For introductory organic chemistry lectures this means understanding basic models like the atomic model (electrons, protons, neutrons) and orbitals and hybridization (if relevant in your class). Why do molecules inherently repel each other, and why do reactions occur nevertheless? What is a chemical reaction? Then, revisit concepts like electronegativity, nucleophilicity and electrophilicity (link), … I might work on a full list of concepts in future.
Understand the functional groups(link): Learn the structures of the most common functional groups in organic chemistry. Sorry, but there is some memorization involved. It’s just like learning a new language: you need to memorize at least new words! For this, you have to understand what a skeletal formula is, and how it differs to normal molecular drawings (next section). Again, I will go into more details in future but here are two easy exercises: =>Get a list of the 15-20 most common functional groups and draw their structures in skeletal formulas. Can’t remember a particular one, like the nitro group and its partial charges? Well, simply draw it 5 times for 2-3 days. You will know all of them in no time. Then, look at them and think about their similarities: For example, a thiol group is like a hydroxyl group (or an alcohol), just that the oxygen is replaced by sulfur which is in the same “group” in the periodic system. An ester is similar to a tertiary amide, just that the -OR group bound to the central carbonyl carbon is replaced by a -NR2 group. Tertiary amides are similar to secondary and primary amides, just that the number of alkyl rests versus hydrogens bound to nitrogen differ. => Google “pharmaceuticals” and check out their structures on Google images. Pick a random pharmaceutical – what functional groups can you identify? Which groups are ones you do not know yet? Do they look similar to any which you know already?
Work your way up the complexity of reactions: 😔 The bad news : There are a literal TON of reactions in organic chemistry. 😊The good news: Thankfully, you don’t have to know all of them, and many of them share the same logic and concepts. Here you should again start easy and build up. => Before trying to understand SN1 reactions or more difficult name reactions, first review acid-base reactions and oxidations/reductions. Then, get into additions. Then, substitutions and eliminations (SN2 and E2 first because they are just one step – then, SN1 and E1). After these, you can start to look at name reactions and complex transformations. For example, if you have are discussing electrophilic aromatic substitutions in class, you need to know how much simpler additions and substitutions work!
Focus on conceptual understanding: It’s normal to memorize things at the start, but really strive to understand the “why” behind reactions and mechanisms. For example: Why does a molecule X react in a substitution – where is its nucleophilic group? Why is the reagent Y an oxidant – which atom has a high oxidation number and gets reduced? Why does the reaction occur regioselectively at this position of the aromatic ring – are there electron-donating or electron-withdrawing substituents somewhere?
Create mind maps: Look at all the reactions you have studies in your class, and try to connect them based on their similarities. Or, connect different concepts: resonance impacts many other factors like acidity of a molecule. How? Electronegativity impacts intermolecular forces like hydrogen bonds and dipole effects. How? Diastereoisomerism impacts melting and boiling points. Why
Create analogies: Whenever you find a concept confusing or can’t remember it, create an analogy. For example, nucleophiles and electrophiles can correspond to rich and broke people, respectively. Electrophiles would like to borrow the electrons of the nucleophiles, creating a strong attraction. The process of the rich lending money to the poor is the nucleophile giving its electrons to the electrophile.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
2. Representation
Master skeletal formulas: Organic chemistry relies on skeletal formulas which omit the explicit indication of carbon and most hydrogen atoms. Remember, each “bend” or end of a line represents a carbon atom, and hydrogen atoms are implied to fill the carbon’s required bonds.
Change your perspective: Molecules can be represented in various ways: Lewis structures, skeletal structures, Newman projections, Fischer projections… Each emphasizes different aspects of a molecule (e.g., stereochemistry). Practice switching between them to strengthen your spatial awareness. => Practice converting between full structures and skeletal formulas until it’s your second nature. Make sure you understand where electron pairs are present! => Convert between Newman projections and skeletal formulas. Are your Fischer projections your enemy? Well, go and solve literally 20 different problems. You will get the hang of it.
Use molecular models to visualize stereochemistry: Stereochemistry is a tricky topic because because it involves 3D thinking. Physical or digital 3D models of molecules can help you see how atoms are arranged in space. => Use online resources like MolView to visualize molecules. Struggling with chirality, enantiomers, and diastereomers? Go to town on the visualization. Rotate, inspect and understand the structures! If you have an university ChemDraw license, use Chem3D!
Practice electron pushing: Organic reactions are governed by electron movement. Curved arrows indicate this electron flow. Whenever you’re learning a new reaction, focus on understanding the arrows, which show how bonds are made and broken. If you can follow the electrons, you’ll have a better grasp on the mechanics of reactions. The same applies to resonance. Pick any molecule of your choice and draw all the different resonance structures. Why resonance structure is the most stable? How does resonance impact the reactivity of the molecule?
Color code drawings for clarity: Use different colors if that helps you. For example, always use red to indicate movement of electrons. Does the reaction involve partial charges? Color code them!
“Play chess” – think several moves ahead: At the beginning, you should write out every single step in a reaction. Once you are at the intermediate level (when you get to practicing retrosynthesis), try visualizing several transformations at once. How would my final product look like if I oxidize the alcohol in the molecule to the aldehyde and perform a Wittig reaction?
3. How to learn organic chemistry
Stay consistent with daily practice: We’ve all been guilty of cramming at some point. However, organic chemistry is best learned through regular practice. Even if you only have 15 minutes, do something related to the course every day—whether it’s reviewing notes, drawing mechanisms, or solving practice problems. The more often you engage with the material, the more familiar it becomes. => Every day, read about one new molecule of your choice. Think drugs against neurodegeneration are cool? Research the structures of Alzheimer’s drugs. What are their functional groups? How are they synthesized? Maybe you are into doing sports? Look at the different types of steroids and performance-enhancing drugs that there are! (Do NOT take them, just read about them…)
Activate your little brain cells: Look, we all are guilty of passively consuming information and entertainment. But instead of just reading textbooks or watching videos, actively engage with the content. Draw out mechanisms as you learn them, explain concepts in your own words, or quiz yourself on reaction types and mechanisms. Basically, anything you are not writing down or creating yourself, you will probably not remember!
Test yourself with practice problems: Organic chemistry is problem-solving heavy, so doing lots of practice problems is essential. Don’t be discouraged by mistakes or getting stuck – this is part of the learning process. The rate-limiting step (see what I did there?) might be how many good problems you can find – you know, ones with proper explanations. The quality of your problems will depend on your luck and teacher. I’m working on a problem book for chemistry myself – but yeah, go to town on Google and YouTube.
Emphasize understanding over memorization: I mentioned this one already – but given we are talking about the learning process here, I wanted to reiterate.
Create your own study materials: Summarize concepts, reactions, and mechanisms in your own words. Create summary sheets, mind maps (see above), or flashcards tailored to your learning style. Experiment and see what works for you! For the things that you will need to memorize (e.g., functional groups, pKa values, named reactions…), you should have a clear approach.
Team up: Organic chemistry is almost always more manageable when tackled with peers. Explain concepts to each other, discuss exercises together, and discuss tricky mechanisms. Your classmates are ahead of you? Great, you can suck out their insights and ask questions. You are ahead of your classmates? Great, help them and in doing so, you will further elevate your capabilities. Are you in university and not against earning an extra buck? Tutor students that are 1-2 years below your level.
Use office hours and ask for help early: If something doesn’t make sense, ask for help right away. Afraid of looking like a moron? Well, the odds are at least half of your class is feeling like morons as well, so don’t feel bad! Visit your professor or teaching assistant during office hours to clarify any misunderstandings. Addressing confusion early prevents small gaps in knowledge from turning into major issues later.
Use multiple resources: Don’t rely solely on one textbook or lecture notes. Different resources may explain the same concept in slightly different ways, and sometimes a different perspective makes things click. Use videos, alternative textbooks, online platforms, and forums to diversify your learning. No access to textbooks? Go to town on LibGen (google it).
Prioritize your effort: As mentioned, building the foundation is key. But once you get into intermediate territory, there are SO many things you could theoretically read about. In the best case, your textbook and/or teacher might have clear objectives or checklists of topics to know. Assess on what you need to work on most, and then go hard.
Is Organic Chemistry Hard?
So, is organic chemistry hard? Honestly – yes, it’s hard.
Is it as hard or terrifying as people make it out to be? No, not really. The great thing is that at some point, every new concept or reaction will be based on something you know already. After enough effort, there will be almost no cases where you really have absolutely no clue of how to approach a problem.
Is it the hardest subject you will study? Maybe, maybe not. Depending on your area of focus, it might come least naturally to you, versus other subjects. If you dislike chemistry and math, physical chemistry will haunt you even more.
To end on a positive: I loved organic chemistry more than any other subject but even I had been struggling with it initially. Resonance? Confusing as hell. Fischer projections? I couldn’t draw a single one correctly. The abbreviation of -Me for a methyl group (-CH3)? I thought it stood for “a generic metal” (lol). I asked A LOT of stupid “questions” (or in many cases did not ask them, which slowed down my progress). What had been a guessing game and brain-dead memorization, became almost second nature.
Whatever your journey in organic chemistry, I hope you can figure out how to make it a bit more bearable and more successful – however you define it: just passing, improving your grade, or really going beyond the class and mastering it.
I’m developing content to help students unlock and practice organic chemistry. If you’re interested, follow along!
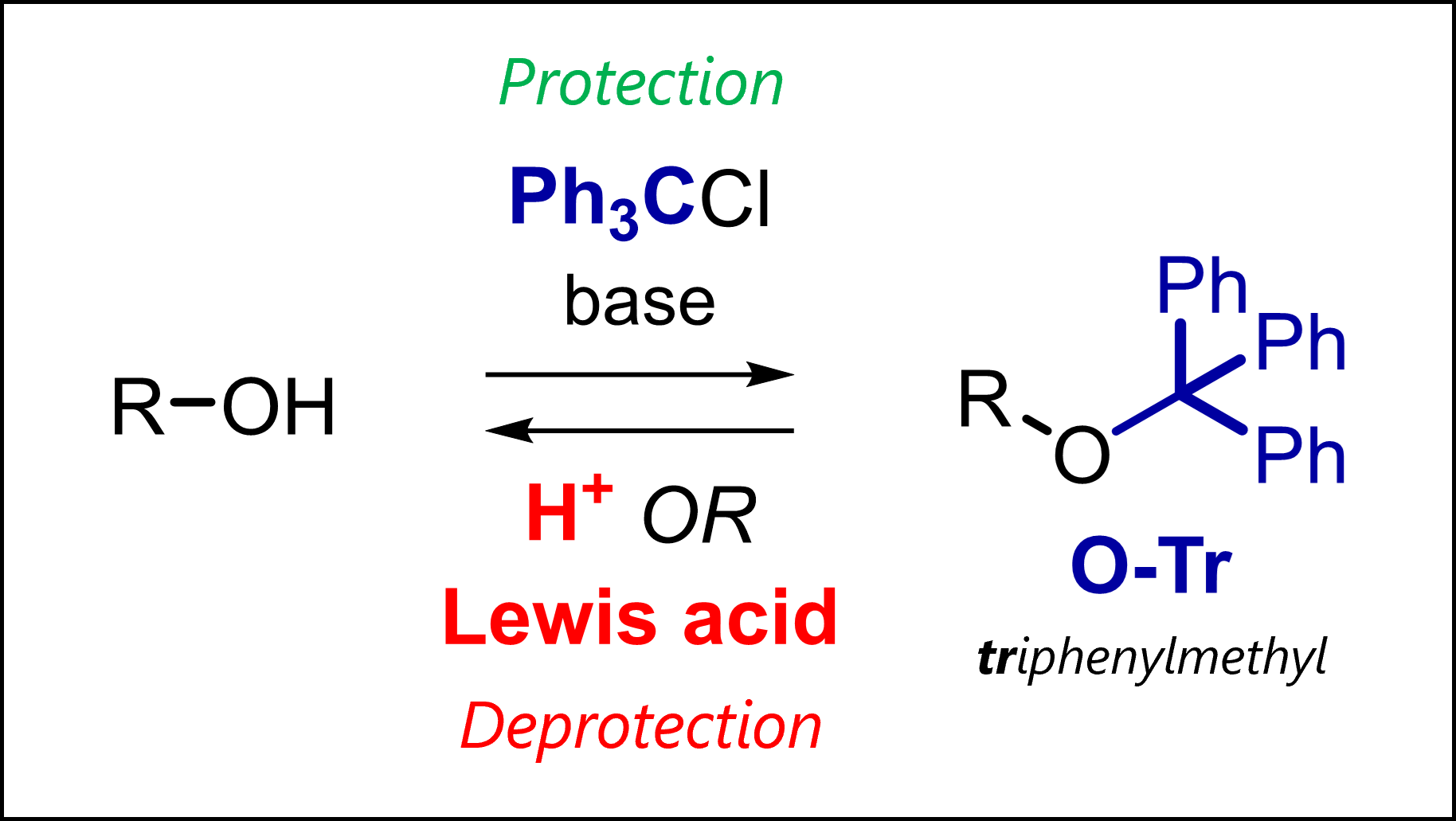
The trityl protecting group protects alcohols in organic synthesis. Tr is deprotected with acids (TFA) or Lewis acids.
The trityl group(Tr / Trt) is an acid-labile protecting group (reminds you of Boc?)! Here we cover “standard” trityl and more advanced versions.
👀 As you see in the 3D model, it’s huge!
Table of contents
What is the Trityl Protecting Group?
Trityl stands for triphenylmethyl, a group most commonly used to protect free alcohols as ethers. As seen with other PGs, amines and thiols can also be protected (as they are also nucleophilic).
Trityl ethers saw most application in carbohydrate chemistry as their hydrophobicity was useful for protection of polar building blocks. In addition, their bulky size allows for selective protection of primary hydroxyls due to sterics. Over time, silyl PGs like TBS have replaced much of the use of Tr outside of carbohydrate chemistry. Nevertheless, we can learn some things by studying it!
Trityl Protection Mechanism
Trityl protection usually uses trityl chloride in pyridine which conveniently functions as a base, capturing the HCl by-product. Added DMAP (4-dimethylaminopyridine) can function as a base but also as a catalyst. You likely remember from other protecting groups (e.g., TBS) or reactions like acylation that DMAP works as a transfer reagent via initial nucleophilic addition to the activated reagent and transfer to our group of interest (not shown above).
Watch out as there are some pages online that imply a direct SN2-like attack of the free alcohol to Tr-Cl. You should know this is impossible; quaternary carbons do not undergo SN2! Instead, the protection proceeds as a SN1 via the stable trityl cation intermediate [1]. The large size allows the selective tritylation of primary alcohols in the presence of secondary alcohols as these react much slower due to steric hindrance.
No surprise, there are other ways to introduce trityl like TrOTf (recall just like for TBS) or trityl-pyridinium tetrafluoroborate, an even more reactive transfer reagent.
Trityl Deprotection mechanism
Trityl is deprotected with Bronsted acids or less commonly also Lewis acids. In both mechanisms, the highly stable trityl cation is a common theme. This should remind you of the Boc group: The trityl cation here is basically a t-butyl cation (acidic Boc deprotection intermediate) on steroids.
Case 1: The deprotection with a Bronsted acid starts with protonation of the ether oxygen. This increases the “pull” on the O-C bond which can fragment to give our deprotected hydroxyl group. The resulting trityl cation is still reactive, so adding nucleophilic scavengers like 2-methyl-2-butene can avoid undesired reactions. By using acetic acid or formic acid, it is possible to deprotect trityl ethers in the presence of TBS ethers. If no sensitive groups are present, stronger acids like TFA obviously work as well.
Case 2: Using Lewis acids like BF3 works in a similar way; coordination to the oxygen lone pair facilitates O-C bond breaking and deprotection. Other Lewis acids like ZnBr2 or MgBr2 can be used for some substrates as well, particularly if two coordination sites are present (e.g., carbohydrates). In these cases, neighbouring group effects with bidentate coordination can be observed.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
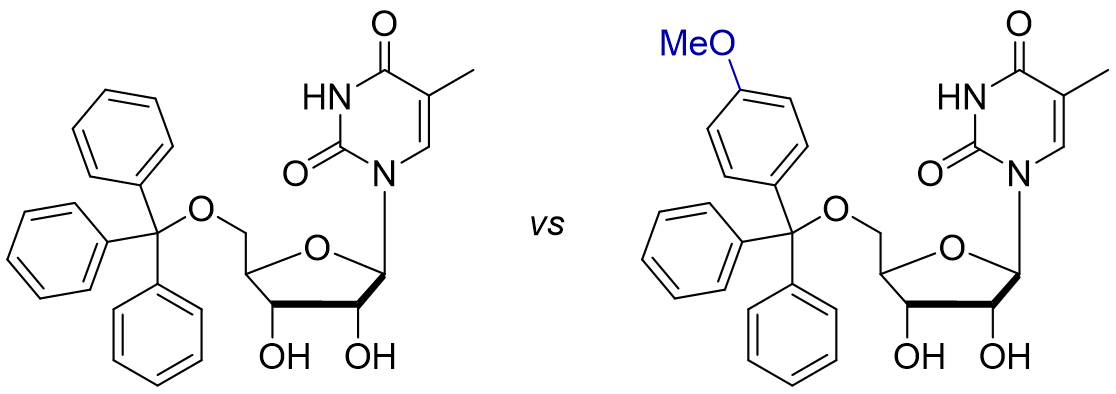
p-Methoxy Trityl Protecting Group
Do you expect the p-methoxy trityl variant to be (more) acid- or base-sensitive?
As a twist on the standard trityl group, chemists have also explored variants such as p-methoxy trityl. A nice synthetic study on nucleotides led to the discovery of such groups – already in 1962 [2]! Their results were as you might expect: By adding a p-methoxy group, we increase stability of the intermediary trityl cation due to the mesomeric electron-donating effect! This makes deprotection easier.
It turned out introducing one methoxy group increased the rate of deprotection by a factor of ten. While standard 5′-trityl-uridine required 48h for complete hydrolysis in 80% acetic acid at room temperature, the mono-methoxy-trityl MMTr group took just 2h! They also developed di- and trimethoxy trityls (i.e., one p-methoxy on each phenyl ring) which cleave in 15min and 1min, respectively. By the way, this change makes initial protection easier too because we also go through the trityl cation.
This di-methoxy DMTr group is one of the most used members of the trityl family due to its reactivity and selectivity for primary alcohols (primarily seen in automated solid-phase synthesis of nucleotides).
A mixture of di-TBS gemcitabine (671 mg, 1.47 mmol) and tritylating reagent (2.94 mmol, 2 equiv.) in dry pyridine (7.3 mL) was stirred overnight at room temperature. Methanol was added to the solution for quenching. After removal of the solvent, the residue was purified by flash column chromatography on silica gel to obtain the title compounds.
Trityl deprotection experimental procedure
Bronsted acid [4]: Compound II (200mg, 0.4mmole) was treated with 3ml of cold formic acid (97+%) for 3 min and then evaporated with an oil pump at room temperature. The residual gum was evaporated twice from dioxane, followed by evaporations from EtOH and Et2O. Finally, the residue was extracted with 10ml of warm H20, the insoluble triphenyl-carbinol was filtered, and the filtrate was evaporated in vacuo. The residual gum was dissolved in EtOH(1ml), dry Et2O (20ml) was added, and the product was precipitated with petroleum ether (30-60°, 10ml) (the gummy precipitate was chilled and scratched to induce crystallization). Recrystallization from the same solvent system gave fine needles of VI.
Lewis acid [5] To a mixture of 4 (2.0 mmol, 994 mg, 1.0 equiv) in CHCl3/MeOH (16 mL/4 mL) was added BF3·OEt2 (4.0 mmol, 0.5 mL, 2.0 equiv) at room temperature. The mixture was stirred at rt for 45 min and was then poured into EtOAc/H2O (100 mL/100 mL). The organic layer was washed with brine (100 mL), dried (Na2SO4), and filtered. After removal of solvent, CH2Cl2 (10 mL) and hexane (30 mL) were added sequentially to the crude product. The resulting solid was filtered and was washed with Et2O/hexane (2/3, 20 mL). The product was dried to give 474 mg of 12 (93%) as a white solid.
Functional groups are the foundation of organic chemistry as they define the structure, reactivity, and properties of organic compounds. Here, we explain what functional groups are (by looking at – wait for it – fruit salad), and introduce the most common functional groups. Did you know that functional groups were at the root of dispute and friendship of two chemistrylegends? Let’s get into it!
What are Functional Groups in Organic Chemistry?
Functional groups in organic chemistry are specific groups of atoms that have characteristic properties (e.g., polarity). Functional groups determine the reactivity of molecules.
Instead of blindly memorizing a long list of functional groups, let us try to understand three key ideas first.
1. Many functional groups are related to each other. For instance, by putting a C=O double bond next to an amine, we get an amide. Others are variants of each other. For example, a sulfur atom instead of an oxygen turns an ether group (R-O-R) into a thioether (R-S-R). When learning these groups, try to find the similarities and differences between them. There are different sub-families of functional groups. For instance, everything with a C=O double bond can be called a carbonyl. Aldehydes, ketones, amides, esters… are all relatives of the carbonyl family! Similarly, primary amines, secondary amines and tertiary amines are all amines!
2. Properties of functional groups are largely independent of the molecule’s broader environment. The hydroxyl group in ethanol behaves the same way as in octanol (even though the latter is much bigger!).
3. Connected to the above, functional groups have an inherent polarity and thus, nucleophilicity or electrophilicity (link). For example, aldehydes are electrophilic on carbon but nucleophilic on oxygen. This baseline polarity of each functional group actually already explains most of the organic chemistry reactions that you will encounter. Interconversion of functional groups can change this natural polarity. For instance, enol forms of carbonyls are not electrophilic at the central carbon anymore, and are nucleophilic at the alpha-carbon!
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
What are Functional Groups – SIMPLY EXPLAINED?
Functional groups are just like different fruits in a fruit salad. Let’s see how the points above match to this analogy.
1. Some fruits are related, like lemons and oranges as they are both citrus fruits. We mentioned carbonyls and ethers above. Another example are primary amines and tertiary amines – very similar but not identical!
2. Whether you catch a strawberry in one fruit salad or another one (these are different molecules in our analogy), they basically taste the same.
3. Lemons are inherently acidic. Well, carboxylic acids are also always acidic. (Sometimes more, sometimes less – due to things like inductive and mesomeric effects.) Just like fruits have characteristic taste, functional groups have characteristic properties. This also nicely illustrates the keto-enol interconversion or even the advanced idea of “Umpolung“. Here, chemists invert the natural reactivity of the original functional group. The key example is conversion of aldehydes to dithianeswhich can be nucleophilic at the carbon (instead of electrophilic)! Using our fruit analogy, by cooking and caramelizing lemons (Umpolung), we can make them more sweet instead of bitter.
History of Functional Groups in Organic Chemistry
Here’s some random background (if you know my content, you will realize I like this type of stuff): As the backbone of organic chemistry, functional groups as a concept have their origin in the early 19th century. At that point, elemental analysis had allowed chemists to determine the molecular formula of most inorganic compounds. These lack carbon-hydrogen bonds and are thus not organic (shocker!). The widespread theory of vitalism suggested that only living organisms can produce organic substances.
Many students know that the German chemist Friedrich Wöhler overthrew this theory by synthesizing urea from inorganic ammonium cyanate in 1828. Less known is the controversy between Wöhler and Justus Liebig: Both of them were doing experiments on inorganic salts that they believed had the molecular formula “AgCNO“. However, Liebig’s salt was a powerful explosive while Wöhler’s was not.
“The obvious conclusion was that one of the analyses must be wrong, and one of the chemists must be a poor analyst! Liebig, pushed by his aggressive character, rapidly accused Wöhler of erroneous results. But Liebig analyzed a sample of the silver cyanate supplied by Wöhler and verified that they were correct. At this point, Liebig openly admitted that he had made a mistake in his initial accusation. And curiously this was the starting point of a friendship and even a scientific collaboration between the two scientists.”
S. Esteban in J. Chem. Educ. 2008, 85, 9, 1201
The legendary Swedish chemist Berzelius (Wöhler’s professor) explained this controversy by proposing the concept of isomers. The realization that constitution of molecules can be different despite having the same atoms paved the way for discovery of all the functional groups we know today.
Common functional groups
Here are the most common functional groups. The bold ones are particularly important as they are invoked to explain some of the basic reactions in organic chemistry, such as nucleophilic substitutions (alkyl halides) or electrophilic additions.
Instead of belabouring basic information you can find everywhere already, I want to draw your attention to three themes:
Functional group relationships: What makes functional groups (in a certain row or across rows) similar? For instance, the first row are C-H hydrocarbon functional groups. Which functional groups are based oncombinations of simpler functional groups? For example, an enone is an alkene that is connected to a ketone. Which functional groups are oxidized/ reduced versions of each other, and which have the same oxidation state? For instance, carboxylic acids are more oxidized versions of aldehydes and ketones, and have the same oxidation state as nitriles.This means nitriles can be converted to acids without reductants or oxidants!
Functional group polarity: What do the red and blue colored atoms correspond to? How does the polarity and reactivity differ across related functional groups? For example, why is the central carbonyl carbon blue in aldehydes and ketones, but not blue anymore in carboxylic acids? Why is the hydrogen of the O-H bond of the carboxylic acid blue but not blue for the C-H in the aldehyde? Should the carbons of alkenes and arenes be marked red or rather blue? What does this depend on?
Functional group geometry: Can you rationalize all bond angles (e.g., alkynes vs. alkenes) and geometries of functional groups? Which parts of them are flat/ in one plane, and which ones are not? For example, why are in esters both the C=O and C-O-R bonds in the same plane? Why do esters prefer the Z-conformer where both C=O and C-O-R bond are facing the same side?
In other posts, we will deep dive into individual functional groups and families to explain properties and most common reactions in more detail. Functional groups clearly also form the basis of protecting groups and their reactivity.
Some final advice: Instead of learning a bunch of facts about 30 functional groups by heart in a brain dead manner, try to work first identify the commonalities and differences from a high level. The names might seem random at first but many of them will make sense once you take a look at what atoms or combinations of functional groups are present!
Did you know that while Coca Cola was getting consumers casually coked up in the late 19th century, chemistry titans were fighting an epic battle around cocaine synthesis? With a Nobel Prize on the line, the stakes were high, cocaine’s structural complexity was high… and everybody else was high too.
Keep reading to learn more about this molecule, as we dive deep into its history and chemistry. Regardless of if you’re a science nerd or amateur, you will learn a ton today – from nice history trivia and tales of careless, chain-smoking chemists, to innovative synthetic strategies that will remain classics in chemistry forever.
This post is purely educational.
Global cocaine use
So, what do the data tell us about cocaine? As one of the most abused substances globally, it’s clearly a huge problem. The number of users has increased faster than population growth. 30% of use comes from the US – and although popularity had been declining for some years, it has unfortunately rebounded more recently. There are no signs a slowdown. The UN even estimates that global use could more than double, in case emerging markets like Africa or Asia would intensify their consumption to similar levels like the Europe or US.
The world’s supply of cocaine originates virtually entirely in South America. The annual manufacture is at record levels of 2000 tons, which is a dramatic uptick from 2014, when the total was less than half as big. On the positive side, interceptions by law enforcement increased faster than production, with some countries seeing 5 to 10-x higher seizures.
Use isn’t everything. The number of overdose deaths involving cocaine has skyrocketed, notably in the US. This is due to the insane increase of synthetic opioids. The light pink line, which excludes these, suggests the US cocaine market has contracted from its peak.
Some researchers have even quantitatively investigated the link of mentions in song lyrics and substance use. The logic here is pretty simple. As rappers increasingly mention baking soda or coco in their songs, they contribute to cocaine popularity and associated deaths. According to the researcher’s statistic model, there is a 2 year lag before this kicks in. At least the use in lyrics seemed to have plateaued in their data.
On a more alarming note, use of even more dangerous crack cocaine has risen in the US and Europe. Compared to powdered hydrochloride salt, this free amine base reaches the brain quicker, making it far more addictive. But what mechanisms of cocaine make it so dangerously enticing for users?
As we will discover shortly, cocaine saw historical use as a local anesthetic. Cocaine stabilizes voltage-gated sodium ion channels in an inactive state. Cells lose their ability to propagate electrical signals, including pain response. Although some FDA-approved cocaine formulations exist, the medical use of cocaine is largely obsolete.
Cocaine’s mechanism of action
What about cocaine’s psychoactive effects? It acts at the synaptic cleft in the central nervous system by blocking certain transporter proteins. These transporters usually mediate the re-uptake of neurotransmitters serotonin, dopamine and noradrenaline. Cocaine mainly prevents reuptake of these agents, causing intense and extended stimulation of the nervous system. On one hand, this overstimulation messes with the body’s reward system. As we’ve seen in our ibogaine video, many psychoactive compounds influence a myriad of targets. The same is true for cocaine. For instance, it also binds opioid receptors – if you want to learn more, check out the literature.
The dopaminergic activity of cocaine makes it addictive, and repeated exposure leads to desensitization. This means people need larger doses to induce stimuli and fend off withdrawal symptoms. This is already problematic, but cocaine and its metabolites are also toxic for many organs such as the liver, brain or kidney. In the cardiovascular system, cocaine leads to an increased heart rate and blood pressure, while decreasing the supply of oxygen to tissues. Remember the sodium channel inactivation? This can change intracardial signals within the heart leading to dysrhythmias. The list of health risks is pretty long, so I hope we all agree that this is not worth it.
Discovery of Cocaine
But since when do we even know about the molecule cocaine itself? Meet the German chemist, Albert Niemann. Like many chemists we will mention today, he is not famous but actually quite the legend. He was in great company, as he was the graduate student of Friedrich Wöhler. This is the guy who synthesized urea – in 1828 from inorganic starting materials. This was a major milestone in organic chemistry, as it dispelled the vitalistic misconception that only living organisms could produce the organic chemicals of life.
Wöhler requested Karl von Scherzer to bring him some coca leaves. This Austrian dude was pretty much living the dream of every gen Z or millennial, as a nice inheritance gave him the financial freedom to quit his printing job and start traveling around. Instead of flying to Bali, von Scherzer participated in the first large-scale scientific expedition of the Austro-Hungarian empire in 1857. Coming home with a stash of coca leaves, he imported some of them into Germany. Just in time for a nice PhD project for Niemann. Although cocaine-enriched extracts were obtained from coca leaves in 1855 already, Niemann was the first to truly isolate and characterize the primary alkaloid. He unsurprisingly gave it the name cocaine and published his research in 1860, earning him his well-deserved PhD.
Niemann was a productive, top notch researcher. In the same year, he described the synthesis of mustard gas from sulfur dichloride and ethylene. Unfortunately, this might have been a case similar to Marie Curie. Niemann correctly noted the toxic properties of this blister agent which as many of you know, saw military use during the first world war. Niemann died shortly after in 1861 due to a devastating lung infection. Maybe, the exposure to mustard gas caused this?
After Niemann’s untimely death, another colleague in Wöhler’s research group concluded his research, determining cocaine’s molecular formula. It might ring a bell for more advanced viewers: this was the guy who coined the Lossen rearrangement reaction. Only thirty years later, Richard Willstätter determined its true structure. This gentleman is the main character of our story.
Early use of cocaine
Before we dive into the chemistry, we should note that the West rapidly embraced cocaine. Initially, it gained acclaim for its local anaesthetic properties, particularly for calming involuntary eye movements during surgery. This was a significant breakthrough! One funny article described it like this: Patients no longer merely endured nerve pain from having their eyes cut; they even chatted pleasantly during their procedures.
Cocaine also found an advocate in Sigmund Freud, the father of psychoanalysis. After using it himself as a stimulant for the smallest of problems, Freud naively recommended it as therapy for various uses, including morphine or alcohol addiction.
His personal positive experiences clearly blinded him, but pharmaceutical companies also paid him to promote their products. Over time, he could not ignore negative consequences such as addiction, leading Freud to eventually reconsider his stance.
Cocaine’s integration into society also took a significant leap with the creation of Coca-Cola in 1886. Originally formulated as a coca wine substitute, Coca-Cola contained coca leaf extract including cocaine, along with caffeine from kola nuts. Due to growing incidence of cocaine addiction, the allrounder power-drink eventually eliminated cocaine from its formula.
Cocaine synthesis: Willstätter’s Starting Material
Let’s talk chemistry. Willstätter didn’t just determine cocaine’s structure – he also completed the first total synthesis of cocaine. Prepare for a wild ride!
His synthesis started from cycloheptanone. But why?
Well, it conveniently brings the largest ring present in cocaine’s structure. Degradation studies by Willstätter, which we will not show in detail, already indicated that cocaine contains a seven-membered ring. To build up the structure back up, Willstätter had to painstakingly functionalize the ring, and create the important amine bridge.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Cocaine synthesis: Functionalizing the core
The first few steps included condensation with hydroxylamine and reduction of the resulting oxime to give the amino group. This new group was eliminated through the Hofmann protocol. It starts with exhaustive methylation to the quaternary ammonium salt, followed by elimination with silver oxide and water. Overall, these four steps gave cycloheptene.
You’re likely thinking – wait a second, why didn’t he just perform a Shapiro reaction to save some steps? Well, this reaction was only discovered in the 1960s, so Willstätter’s armamentarium of chemical reactions was limited.
One double bond is nice, but as you will see, we actually need three of them. Bromination gave the dibromide, temporarily removing the alkene. Addition of dimethyl amine led to substitution and elimination, giving a double bond. Before you get excited, this is not yet the amine for the tropine bridge. Rather, another Hofmann elimination gave the diene. Because this was such great fun, another bromination and elimination with quinoline as a weak base gave cycloheptatriene.
Now, kinda going a step backwards, the triene was reacted with hydrogen bromide. Due to the intermediary formation of the allylic cation, this regio-isomer is preferred as opposed to the other addition product. The newly introduced bromide group was substituted with dimethyl amin. At last, this is the amine that we need in cocaine. The next step was basically why the triene approach even worked. Reduction with sodium metal in ethanol reduced just one of the double bonds, resulting in a single isomer with the surviving alkene situated on the other side of the cycloheptane. Willstätter himself commented himself that selective reduction is strange. The last preparatory steps were a bromination of the remaining olefin to the trans-dibromide which also formed the ammonium salt. This was neutralized with sodium carbonate, regenerating the electron pair of the amine. This set up the critical step of the synthesis.
Intramolecular alkylation en route to cocaine
So, our molecule now has a potent nucleophile – the amine – as well as electrophilic carbon-bromide bonds. In the energetic ground state, the groups don’t get close so there’s no reaction. However, cooking things up in ethanol provides sufficient flexibility for the ring to distort and get the amine close enough for a transannular SN2 reaction with the anti-bromide. As we’ve explained in detail in a previous video, the reaction proceeds through back side attack and a pentacoordinate transition state. This synthesis is racemic – you can check for yourself that an inverted configuration at the amine leads to the same product as it simply kicks out the other bromide instead.
The product is obtained in roughly 30% yield and from what I could tell reading Willstätter’s work, it looks like he actually coined the term “intramolecular alkylation”. For 21st century chemists, this reaction looks simple, but for 1901, this is really insane foresight. Having created the tropane skeleton present in cocaine, we still have to demethylate the ammonium group, and functionalize the two ring positions.
Synthesis of Tropidine and tropine
To this end, elimination of the remaining bromide gave the double bond, setting up upcoming functionalizations. Then, the ammonium ion was converted to the neutral amine. Earlier, we’ve had the protonated amine which is why base was sufficient for neutralization. However, now we are looking at a tetra-alkyl ammonium. So, chloride anion exchange and heating removed one of the methyl groups via intermolecular SN2 reaction.
How do we wrap up the synthesis? Well, instead of the alkene, we need an ester and a hydroxy group that we can benzylate. High school chemistry tells us that we can easily add water to double bonds, but in this case, a direct hydration was impossible. Instead, this conversion took two steps. First, exposure to hydrogen bromide gave the secondary bromide. Under acidic conditions, hydrolysis of the bromide gave the alcohol.
Willstätter and Ladenburg: Two Titans Clash
This step took quite some experimentation to get right, and actually it was a source of dispute between Willstätter and Albert Ladenburg, another German chemist. Ladenburg had claimed the direct synthesis of the alcohol under cold hydrobromination conditions more than a decade earlier. Willstätter criticized it, claiming year-long experiments did not replicate his findings.
Very sneakily, Ladenburg updated his experimental procedures in later publications – saying they were included insignificant modifications. However, Willstätter saw this as a copy of his own research. His procedure strictly required acidic conditions and heat. To him, Ladenburg was hoping to cover up unsuccessful, fabricated findings without drawing too much attention.
This was not the end – within the same year, Ladenburg retaliated, accusing Willstätter of defaming him by using cheap tricks. He claimed to had repeated the synthesis, not specifying what small amounts of tropine mean, and again downplaying his modifications. I checked his OG report and while correct that he explicitly mentioned use of increased temperatures, he did not add acids or water. You would not call me crazy when I say that Willstätter’s conditions in blue look pretty important to enable hydrolysis of the bromide.
I’m sparing you the details – but this story really shows how challenging old school chemistry was. Good luck proving that someone did or did not synthesize a structure when the characterization method was doing random characterizations of platinum salts. Basically, Ladenburg was like trust me bro, I know these platinum salts very well.
To him, this was bullet-proof evidence, and he concludes his position as arrogantly as he could. I didn’t check subsequent correspondence but a web-text on Ladenburg noted that he had quite some personal hostilities, so I’m not surprised.
My vote goes to Willstätter. Ladenburg, again in peak 1900s style, used the low volatility of his putative tropine product as an argument to prove its identity. Turns our whatever Ladenburg had, it was not tropine, as Willstätter showed that tropine is in fact volatile. Ladenburg hid his sneaky changes to such a degree that even a gentleman reviewing and writing about the procedure did not catch the changing method. Although you might not like Ladenburg knowing this, we have to give credit where credit is due.
He was in fact the first chemist to synthesize an alkaloid. This means he is one of the founding fathers of total synthesis. Starting from 2-methyl pyridine, he synthesized coniine – a simple but toxic alkaloid – in two steps. First, a condensation reaction delivered allyl pyridine and second, complete reduction with sodium gave the target. Given the condensation required 250 °C, you won’t be surprised that the yield was “pretty bad” as Ladenburg put it – 45g out of 1kg of educt. Just like today’s synthetic chemists struggling in total syntheses, he had to perform multiple reaction cycles by recovering unreacted starting material. Ladenburg also managed to separate the enantiomers through separation of diastereomeric tartrate salts.
Cocaine Synthesis: final Steps
Back to cocaine. The conversion of tropidine to tropine was actually the missing link. Willstätter himself proudly concluded his report that this step completed the total synthesis of various alkaloids, including cocaine. The remaining steps were already known so the synthesis was not discovered linearly. Let’s see how we can wrap things up. Of course, the newly formed hydroxyl group could be benzylated but that wouldn’t help us with the missing ester group.
Thus, the hydroxyl group was first oxidized to the ketone which allowed alpha-functionalization. Nucleophilic addition of the enolate with CO2 creates the last important C-C bond. The addition is diastereoselective for the axial product, opposite to the rest of the tropine skeleton.
Instead of direct esterification, the ketone was reduced first. You might be surprised, wondering why the hydride now has added from the bottom face? Don’t forget the bunny ears! In the equatorial product, the hydrogen enjoys almost perfect alignment with one of the oxygen’s lone pairs, leading to a short and strong intramolecular hydrogen bond. However, the reported yield was low with no info on product ratios, so we can’t really say what was thermodynamically or kinetically favored.
The last steps were pretty simple. First, the methyl ester was completed through acidic Fischer esterification. Finally, addition of the activated reagent benzoic anhydride introduced the benzyl group, concluding the first total synthesis of cocaine. All in all, Willstätter’s effort spanned roughly 23 steps, and the net yield must have been horrendous.
This wouldn’t be a century-old tale without the chemists trying their synthetic cocaine, noting a bitter taste and characteristic feeling on the tongue. Willstätter elucidated and/or synthesized other important molecules, including proline and cyclooctatetraene. He received the Nobel Prize in 1915, primarily actually due to this research on the structure and function of chlorophyll.
Robinson’s Cocaine Synthesis (1917)
Willstätter’s total synthesis was remarkable feat of skill and perseverance, with the majority of the effort focusing on the synthesis of tropinone. Unfortunately, there’s always another chemistry legend out there getting ready to ruin your career. We’re talking about Robert Robinson, well known for his own Nobel Prize and coining key chemistry concepts we still use today – such as curly arrows representing electron movement.
Robinson published research in 1917 that made Willstätter’s signature efforts look pretty outdated. Robinson didn’t hold back, noting that the method was so complicated that he didn’t even bother to recall it in detail. His argument was that the long and low-yielding synthesis did not represent an economically workable alternative to natural sources.
Robinson’s breakthrough finding was a one-pot reaction of three components, giving tropinone in an impressive 42% yield. If haven’t seen this one before, feel free to pause the video right now, and think about a potential mechanism for this reaction.
The reaction starts off with condensation of the nucleophilic methylamine to one of the electrophilic aldehydes. After loss of water, the intermediary imine is still nucleophilic and can form a ring by a second addition – turning it into a positively charged electrophile. The enol tautomer of the acetone dicarboxylate steps in and adds to the iminium in an intermolecular Mannich reaction.
Due to the carboxylate groups, the acetone reagent functions as a di-anion equivalent. After the addition, we can have a decarboxylation of one of the groups, and reprotonation. As the nitrogen regained its electron pair after the first Mannich reaction, it can kick out the adjacent hydroxyl group, creating yet another electrophile that is ready to undergo a second, now intramolecular Mannich reaction. A second decarboxylation affords tropinone in a process that is, as we would all agree, much simpler than Willstätter’s approach.
Noyori’s Cocaine Synthesis (1974)
Note that by the early 20th century, the use of cocaine was peaking. Newspapers were filled with ads promoting the drug, and people were using it either recreationally or ironically, to overcome morphine addiction. Thousands of deaths from cocaine abuse prompted the US to regulate the drug in 1914. This removed cocaine from over-the-counter remedies and consumer products.
How did the chemistry evolve? For the sake of science of course, many chemists tried to synthesize tropane alkaloids in a more efficient manner. One elegant approach was described by Noyori in 1974, who is well known for his discovery of asymmetric hydrogenations. This won him the Nobel Prize together with Knowles in 2003. 1974 was actually the year when Noyori and co-workers initiated the synthesis of BINAP diphosphane.
Just like Robinson, Noyori attempted to simplify the synthesis of tropinone – the intermediate that Willstätter only accessed painfully over dozen steps. He disconnected the same bonds but instead of Mannich reactions, he employed a cycloaddition to link the ring. Don’t blink, because this one is also very short.
The reagent diiron nonacarbonyl is a reactive source of iron(0) which can reduce the tetrabromoacetone to an oxyallyl intermediate. A [4+2] cycloaddition with a pyrrole creates the tropane structure. If you’re confused by the charges, the addition does form a positive charge on the central allyl carbon but the negative charge on the oxygen regenerates the ketone. Two reductions gave tropine – first, the superfluous double bond and bromides were removed with hydrogenation, and second, the ester was reduced to the methyl group.
Why even have these extra bromo groups and ester in the first place? Well, initial attempts with dibromo acetone instead of tetrabromo acetone as a precursor did not form the oxyallyl intermediate. Similarly, the direct use of N-methyl pyrrole led to electrophilic substitution products on the pyrrole, as opposed to cycloaddition. It makes sense use of the ester deactivates the pyrrole, taming its reactivity for substitutions. Noyori nicely overcame these challenges, but just like all other syntheses, the value of this synthesis was primarily in the chemistry, rather than allowing easier access of cocaine.
Cocaine Biosynthesis
As a side note on biochemistry, the biosynthesis of cocaine only fully solved in late 2022. This research showed that the biosynthesis of tropane alkaloids from different plant orders – here in green and pink – uses unique enzyme classes but arose independently at least twice during the evolution of land plants. Similar to Robinson’s one-pot tropinone synthesis, we have an electrophilic iminium which reacts with an activated nucleophile. However, compared to Robinson’s electrophile, this iminium is not doubly activated. This means that there is no dual addition and decarboxylation. Instead, there is enzyme magic to oxidize and cyclize the tropane ring. If you are interested in biosynthetic pathways, you can check out the clever isotope-labelling experiments which elucidated some pieces of the puzzle already in the mid-20th century.
Cocaine synthesis via Engineered Tobacco?
Many organic chemists think biochemistry is boring but check this out. By understanding biosynthetic pathways, scientists have recently genetically engineered tobacco plants to produce two new enzymes in their leaves.
This is far too complex for large scale criminal synthesis of cocaine, but pretty cool. Agrobacteria can transfer and insert parts of its DNA into plant cells – giving tobacco the tools to synthesize cocaine. In a funny parallel to the chemical synthesis of cocaine, the last step is a benzoylation. Tobacco produces endogenous benzoic acid, but the plants were treated with additional benzoic acid to further double the cocaine yield – which is way higher than I would have predicted.
Congrats, you now probably know more about cocaine than everybody else you know. Thanks for following my content, and see you in the next one!
If you are interested in the academic synthesis of psychedelics, check out the discussion of ibogaine, psilocybin, MDMA or THC-P.
Cocaine synthesis references
– UNODC, Global report on Cocaine 2023 (United Nations publications, 2023) – Estimating the incidence of cocaine use and mortality with music lyrics about cocaine | npj Digital Medicine 2021, 4, 100 – Cocaine: An Updated Overview on Chemistry, Detection, Biokinetics, and Pharmacotoxicological Aspects including Abuse Pattern | Toxins 2022, 14, 278 – DARK Classics in Chemical Neuroscience: Cocaine | ACS Chem Neurosci 2018, 9, 2358 – Cocaine Use Disorder (CUD): Current Clinical Perspectives | Subst Abuse Rehabil. 2022; 13: 25 – Ueber die Einwirkung des braunen Chlorschwefels auf Elaylgas | Justus Liebigs Annalen 1860, 113, 288 – Synthese des Tropidins | Ber. Dtsch. Chem. Ges. 1901, 34, 129 – Ueber die Umwandlung von Tropidin in Tropin | Ber. Dtsch. Chem. Ges 1902, 35, 1870 – Umwandlung von Tropidin in Tropin | Ber. Dtsch. Chem. Ges 1902, 35, 2295 – Synthese der activen Coniine | Ber. Dtsch. Chem. Ges 1886, 19, 2578 – Richard Willstätter and the 1915 Nobel Prize in Chemistry | Angew. Chem. Int. Ed. 2015, 54, 11910 – A synthesis of tropinone | J. Chem. Soc., Trans., 1917,111, 762 – New, general synthesis of tropane alkaloids | JACS 1974, 96, 3336 – Elucidation of tropane alkaloid biosynthesis in Erythroxylum coca using a microbial pathway discovery platform | PNAS 2022, 119, e2215372119 – Discovery and Engineering of the Cocaine Biosynthetic Pathway | JACS 2022, 144, 22000 (not 21809)
Ever struggle with how to identify if a group is a nucleophile vs. electrophile? If you are in a rush and don’t care about learning chemistry (bruh), the first sections got you. I recommend you try to truly understand the concept as it’s arguably the most important skill in organic chemistry.
Table of contents
Nucleophiles (nucleus-loving) are neutral or negatively charged species that donate high energy electrons to form new bonds with electrophiles (electron-loving), which are neutral or positively charged species that can easily accept electrons
Nucleophiles are Lewis bases (e.g. NH3); electrophiles are acids (e.g. H+, BF3)
The chemical bonding occurs between the highest occupied (HOMO) of the nucleophile and the lowest unoccupied molecular orbital (LUMO) of the electrophile
Single atoms, sigma-bonds and pi-bonds can be nucleophilic or electrophilic – and the same molecule can contain both (one part nucleophilic, one part electrophilic)
Nucleophiles and electrophiles are complementary, they react with each other. You will never see a reaction where two groups react with each other as nucleophiles!
Level 0: Chemical bonds are like … financial transactions?
Chemistry is like finance! Simply swap electrons (chemical currency) for money.
If you’re a student, you’re likely broke(you are in need of, and easily accept money => you are electrophilic). You go to your parents to borrow some money for a dinner (they can donate money => they are nucleophilic). By getting money and the food, you get happier (you are energetically stable). Your parents are also stoked they can spend some time with their favorite child (they are also energetically stabilized).
This explains how we draw curly arrows, representing electron movement: There are no electrons at the electrophile (it is broke). Instead, the electron donation arrow starts at the nucleophile and points to the acceptor, the electrophile.
Level 1: Nucleophile vs electrophile
You have two options on how to remember which is which: 1. Not recommended: You force-memorize some analogy like above with no brain cell activation, setting you up for nice failures in organic chemistry exams
2. Recommended: You just use your brain and simply remember that “phile” means liking or loving (if you struggle to remember, you know a highly criminal term…)
Nucleophile
Electrophile
P*d*phile
What it has
Has more than enough electrons
Has some sort of positive polarization
Problems, chocolate & puppies
What it wants
Wants to share its electrons (likes positive charges)
Wants to accept other electrons (likes electrons)
Kids
Charge
Neutral or negative
Neutral or positive
Prison sentence
Orbitals
High-energy occupied
Low-energy unoccupied
Orbits around kids and playground
Acid or base
Lewis base
Lewis acid
Puts them in his base(ment)
The electrons that a nucleophile donates form the new bond with the electrophile. A simple example is the protonation of water. Which is the nucleophile vs electrophile?
Reviewing the more complex mechanism for the hydration of formaldehyde, you will realize that: 1. Specific atoms or also bonds can have nucleophilic or electrophilic behavior For example, in the first step, the carbonyl pi-bond is the electrophile. In the second step, the proton H+ is the electrophile.
2. For reactions with multiple mechanistic steps, there can be many different nucleophiles and electrophiles For example, after the first addition to the carbonyl, the previously nucleophilic water molecule turns into a cationic intermediate which is now electrophilic (which is why it reacts with another nucleophilic water in a deprotonation)
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Level 2: Types of nucleophiles
How do we identify electron donors and electron acceptors? There are threecategories for both. It’s easier for nucleophiles, so let’s start there.
To donate an electron pair, you need to have an electron pair somewhere(duh). In our analogy, you need to find the bank vaults that are filled with cash(electrons)!
1A) Lone electron pairs, e.g., water H2O or ammonia NH3 (neutral nucleophile) Reaction example: Protonation of neutral bases 1B) Negative charges (also electron pairs), e.g., hydroxide OH– or cyanide CN– anions Reaction example: Basic hydrolysis of an ester 2) Bonding pi-orbitals, most notably C=C double bonds, e.g., alkene or aromatic ring Reaction example: Bromination of alkenes, electrophilic aromatic substitution 3) Bondingsigma-orbitals with highly electropositive atoms, e.g., methyllithium Li-CH3 Reaction example: Carbonyl reduction with LiAlH4, organometallics
This should make sense. These are the only “ways” you will ever find electrons in a molecule. Either as an unbound electron, in a pi-bond, or in a sigma-bond. This corresponds to the idea of highest occupied molecular orbitals (see below).
Level 2: Types of electrophiles
What about electrophiles? Two categories are similar, and one is different.
To accept an electron pair, you need to have an empty orbital somewhere (duh). Here, we need to find the empty wallets that are eager to get filled with money!
Like nucleophiles, we have pi- and sigma-bonds as acceptors (as they have empty orbitals that can be filled with electrons). However, instead of electron pairs for nucleophiles, we need to look for empty orbitals on single atoms. Think of the empty orbitals like filled purses
1A) Positive charges representing an empty p orbitals, e.g., proton H+ Reaction example: Protonation of any base 1B) Neutral molecule with empty p orbital, e.g., Lewis acids like BF3 or AlCl3 Reaction example: Friedel-Crafts acylation 2) Pi-bond next to electronegative/ stabilized system, e.g., carbonyl Reaction example: Aldehyde hydration, conjugate addition 3) Sigma-bond to electronegative atom, e.g., methyl iodide CH3-I Reaction example: SN2 reaction, bromination of alkenes
For both of these categories, I always represented the nucleophile as “Nu-” and electrophile as “E+”. But any combination of these categories works – e.g., an nucleophilic pi-bond can attack an electrophilic pi-bond!
If you know a fair share of reactions, try to think about additional examples for each!
Level 3: Orbitals
Let’s get to the bottom of this (without me writing another text book on orbitals).
Remember that reactions take place if molecules…
1. Overcometheir electronic repulsion via charge attraction and/or orbital overlap (all reactions have activation energies) 2. Have orbitals of appropriate energyto interact (if orbital energies are too different, there is no energy gain from bonding) 3. Orient in a geometry that allows their orbitals to overlap in a bonding interaction (if a key can’t get into a lock, the door can’t be unlocked)
Point #2 is critical: energy levels of the nucleophile and electrophile orbitals. The strongest stabilization comes from interacting orbitals with similar energies.
All molecules have many low-energy, unfilled orbitals and high-energy, empty orbitals (illustrative grey orbitals). We can ignore all because the energy differences between them are too large. The most relevant interaction will be the one between the lowest-energy unoccupied (LUMO) and highest-energy occupied molecular orbital (HOMO).
The higher-energy a HOMO is, the easier it can donate electrons into LUMOs. The lower-energy a LUMO is, the easier it can accept electrons.
This picture explains the types of electrophiles… 1A + B) Empty orbitals can be LUMOs (neutral or positively charged atom) 2) Pi-bonds always have an empty antibonding pi-star MO which can be a LUMO 3) Similarly, sigma-bonds have an antibonding sigma-star MO which can be LUMO
… and types of nucleophiles: 1A +B) Lone pairs can be HOMOs (neutral or negatively neutral charged atom) 2) Pi-bonds always have a filled bonding pi MO which can be a HOMO 3) Similarly, sigma-bonds have a filled bonding sigma MO which can be a LUMO
Recap: Nucleophile vs electrophile
By now you should be able to identify a nucleophile vs electrophile, and know the different types that exist. As an exercise, review reactions you already know or that I discuss on my channel, or functional groups (e.g., protecting groups) to apply your learnings.
There are more nuances and explanations which are not digestible in a single post. So, future posts will explain things like: – What are the most common nucleophiles and electrophiles? – How does conjugation to electron-donating or -withdrawing groups influence nucleophilicity or electrophilicity?