The main protecting groups for alcohols are ethers, silyl ethers and acetals. Here’s an overview of their structures, protection and deprotection conditions.
For more details on some of these protecting groups, check out these articles. For each group, we explain the context, mechanisms for protections and deprotections, 3D models and examples (ranging from easy to hard). O-PMB | O-Trityl | | O-TBS | O-MOM | O-THP | O-MEM | O-SEM
When are Alcohol Groups Protected During Organic reactions?
This might be an obvious question we explain here already: Why do we need protecting groups to begin with?
It’s all about getting what you want, while avoiding what you don’t want.
Protecting groups are temporary shields for reactive functional groups to prevent side reactions and maximize chemoselectivity of reactions.
When we modify more complex molecules, we might want a specific functional group to react selectively while preserving other hydroxyl groups presentin the molecule. The synthesis of carbohydrates (and other natural products, like oxygenated terpenoids) thus often requires protecting groups for alcohols.
The issue comes from one of the first reactivities we learn about in organic chemistry: the chemistry of water (H2O) and alcohols, like methanol (CH3OH). The “-OH” hydroxyl group is a nucleophile and can interfere with reactions of other functional groups.
It is somewhat possible to selectively functionalize some groups without protecting groups, for instance by leveraging relative reactivity due to steric hindrance. Primary hydroxyl groups react faster than secondary hydroxyls. However, for the assembly of complex molecules with dozens of reactive functional groups, protecting groups are basically impossible to avoid.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Let me know if you would like additional summaries of protecting group classes (e.g., for amines etc.). If you’re interested in practice problems in organic chemistry, check out the free problem sets.
The Alloc protecting group is used to protect amines. It is removed with catalytic Pd(PPh3)4 and thus orthogonal to many PGs.
🫡 Here’s what you’ll learn in this article:
Table of contents
👀 BTW, notice how in the 3D model, the allyl group is not planar with the carbonyl!
What is the Alloc Protecting Group?
The Alloc protecting group is related to the Allyl protecting group and orthogonal to almost every other protecting group out there.
It might seem complicated, but it’s just a fancy version of a carbamate protecting group based on two structures: 1) A carbamate which if present in its free form (-OH), can decarboxylate to release the original amino group. Other carbamate protecting groups we’ve seen like Boc, Cbz or Fmoc work this way. They just have a different “trigger” group. 2) An O-allyl group that is activated towards nucleophilic attack of palladium(0) to form of allyl-palladium complexes. Instead of e.g., a tBu group present in Boc which gets “triggered” by acid, this one is triggered by Pd(0)!
Due to the mild deprotection conditions, the group has seen significant application in the synthesis of complex peptides and carbohydrates as a solid alternative to e.g., Boc.
Alloc Protection Mechanism
Alloc protection is trivial and follows the same logic like other carbamates: Nucleophilic attack of the amine to some sort of activated Alloc reagent, typically AllocCl (allyl chloroformate – so like CbzCl for Cbz) or Alloc2O (diallyl dicarbonate – like Boc2O!).
Exemplary conditions are i) AllocCl, pyridine in THF; ii) AllocCl, DMAP, NEt3 in CH3CN; iii) Alloc2O in dioxane, H2O or in CH2Cl2; iv) Alloc-OSu, NEt3, CH2Cl2
Alloc deprotection mechanism
As already alluded to, the magic of this group is in the activated allyl group (given it’s connected to the oxygen of the carbamate). The deprotection is a catalytic cycle, initiated by coordination of Pd(0) and oxidative addition to form an allyl-palladium(II) complex.
The carbamate ligand can dissociate from the complex and decarboxylate to give our desired deprotected amine (again, the same logic as we saw with Boc, Cbz and Fmoc).
But how do we regenerate our catalyst Pd(0) and get rid of the allyl group? There are two options: 1) Nucleophiles can attack the complex and transfer the allyl group, reducing Pd(II) to give Pd(0). Morpholine is one of the key allyl transfer reagent in the text books, but other amines (Me2NH•BH3), C-H acids (e.g., dimedone, barbituric acid)… can be used. 2) Hydride donors can reduce the allyl group via reductive elimination, giving butene. Silanes (such as PhSiH3 / phenylsilane) are common but other hydride donors such as formic acid, SnBu3H (tributylstannane) or sodium borohydride (NaBH4) exist.
If no additional allyl transfer reagent / scavenger would be added, the cycle would not be closed and we would see undesired allylation of our deprotected amine (because it’s a nucleophile).
If you are crazy but want some of that OG E. J. Corey chemistry swag [1], you can also use Ni(CO)4. This compound is extremely toxic and highly volatile… but why would you if you can use palladium?! Fun fact: The name of Corey’s co-worker in the paper [1] is “Suggs” – so yeah, working with nickel carbonyl suggs!
Pd(0) is unstable. To not prepare it fresh, it can be formed from Pd(II) precursorcatalysts like Pd(PPh3)2Cl2 and reductants such as silanes. Not all Pd(0) reactions start with Pd(0)!
Examples of Alloc PROTECTION in Organic Synthesis
This first example [2] shows the orthogonality of Alloc – here, with Fmoc and a methyl ester. In this case, the borane-dimethylamine complex is used as a nucleophilic allyl transfer reagent.
Our second example [3] is from the total synthesis of antillatoxin. This marine natural product was isolated from some exotic cyanobacteria and, in addition to being toxic to shrimp (lol), might have interesting bioactivity (antiproliferation of cells via inhibition of tubulin polymerization).
You’ll see that two allyl groups were deprotected in one step – one from an amino group, and one from an ester. This set up the final intramolecular cyclization step to form the lactam (cyclic amide) in the product.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
That’s All(oc) for this article! (ok, bad pun…) Feel free to check out my other articles on protecting groups, my page or my videos!
Alloc Protection experimental procedure [4]
“A mixture of amine (0.0842 mmol), NaHCO3(44 mg, 0.53 mmol, 6 equiv), THF (3 mL), and H2O (3 mL) at room temperature was treated with allyl chloroformate (28 μL, 0.26 mmol, 3 equiv). The reaction mixture was stirred at room temperature for 12 h, extracted with EtOAc (200 mL, 100 mL), and the combined organic layers were washed with saturated aqueous NaCl (200 mL), dried over Na2SO4, and concentrated in vacuo. Column chromatography provided 38 (48.8 mg, 87% over 2 steps) as a white foam.”
Alloc deprotection experimental procedure [4]
“A stirred solution of 40 (8.61 g, 8.2 mmol, 1.0 equiv) in CH2Cl2 (82 mL) at 0 ̊C under Ar was treated with PhSiH3 (7.1 ml, 57 mmol, 7.0 equiv) followed by Pd(PPh3)4 (0.95 g, 0.82 mmol, 10 mol %). The reaction mixture was stirred at 0 °C for 1 h and concentrated under reduced pressure. Column chromatography provided the semi-pure amine (7.79 g) as a yellow solid.”
Alloc Protecting Group References
General: P. G. M. Wuts, T. W. Greene: Greene’s Protective in Organic Synthesis (Wiley)
[1] Cleavage of allyloxycarbonyl protecting group from oxygen and nitrogen under mild conditions by nickel carbonyl | E. J. Corey, J. William Suggs | J. Org. Chem. 1973, 38, 3223
[2] P. J. Kocienski: Protecting Groups (Thieme)
[3] Total Synthesis and Revision of Absolute Stereochemistry of Antillatoxin, an Ichthyotoxic Cyclic Lipopeptide from Marine Cyanobacterium Lyngbya majuscula | Fumiaki Yokokawa, Hideyasu Fujiwara, Takayuki Shioiri | Tetrahedron 2000, 56, 1759
[4] Next-Generation Total Synthesis of Vancomycin | Maxwell J. Moore, Shiwei Qu, Ceheng Tan, Yu Cai, Yuzo Mogi, D. Jamin Keith, Dale L. Boger | J. Am. Chem. Soc. 2020, 142, 16039
The SEM protecting group is used to protectalcohols and amines. SEM is removed with fluoride(F–) or acid(H+).
2-(Trimethylsilyl)ethoxymethyl (SEM) is an acetal-type PG. 🫡 Here’s what you’ll learn:
Table of contents
👀 No surprise, SEM is related to MEM and the simpler MOM protecting group. But note the TMS!
What is the SEM Protecting Group?
The SEM protecting group is essentially a combination of the MEM group and a TMS group. The presence of silyl group means that fluoride can play a part in deprotection as well.
When attached to an alcohol, it forms a much less reactive acetal. However, just like other similar protecting groups, it can also be added to other nucleophiles like amines. SEM is stable under various conditions, including bases, reductants, organometallic reagents, oxidants and mild acids.
The SEM group was invented in 1980 by Lipshutz and Pegram [1], so just a few years after introduction of MEM by E. J. Corey.
SEM Protection Mechanism
The protection is MOM and MEM all over again. 1. Option: Treatment with SEM chloride and DIPEA (N, N-diisopropylethylamine) or another weak base. Deprotonation occurs after nucleophilic attack. 2. Option: Treatment with a strong base like NaH (KH, n-BuLi, …)and SEMchloride. Experimentally / in the lab, only base is added to the alcohol first – and only after some time (e.g., 1h to ensure the alkoxide is formed), SEMCl is added.
Again, note the activation and higher reactivity of such alkylating agents due to the adjacent oxygen.
SEM deprotection mechanism 1: Fluoride (F–)
The presence of silicon in SEM allows for deprotection with fluoride anions (high thermodynamic affinity for the very strong Si-F bond). These mechanisms proceed via formation of the pentavalent siliconate intermediate.
The siliconate is unstable and can trigger a beta-elimination decomposition. This releases three neutral molecules: TMSF, ethylene and formaldehyde. We can just draw everything in one single step and take a proton from the solution (under acidic conditions like HF, the oxygen might be protonated already prior to decomposition).
The benefit of fluoride is that it the conditions are often orthogonal / compatible with many functional and protecting groups. However, compared to ordinary silyl ethers (e.g., TMS), SEM deprotection tends to require higher temperatures, longer reaction times, or in some cases the use of additives (HMPA).
Exemplary fluoride deprotection conditions for SEM are i) TBAF, DMF; ii) HF, MeCN; iii) LiBF4, (MeCN-H2O).
SEM deprotection mechanism 2: Bronsted Acid (H+)
Though less preferred than fluoride-mediated deprotection, acidic hydrolysis also works for SEM. This is another parallel to MOM, MEM and THP. There are more mechanistic options , but the direct path is probably the most likely one.
i) Direct path: The oxygen of our protected alcohol is protonated and achieves the deprotection in the simplest manner.
ii) Indirect path: The other ether oxygen is protonated, and goes via the hemiacetal intermediate (formaldehyde is lost in the solution or upon work-up). This is what we’ve seen for MEM or MOM.
iii) Beta elimination: What we’ve seen during the fluoride deprotection, but now just in the acidic variant.
What about relative stability? According to Kocienski [2], SEM is more labile than MOM and MEM under acidic conditions. (Obviously, fluoride does not remove MOM). On the other hand, Greene [3] notes that SEM are very robust groups and often difficult to remove.
The truth is probably more the latter. Here is an example from the total synthesis of taxol by Kuwajima [4].
“We also investigated the Birch reduction of derivatives of 25a with various protecting groups on the C2-OH: reaction of the di-tert-butylsilylene derivative 25b induced C2−O bond cleavage predominantly (eq 3). Use of the C2-O-SEM substrate 25c gave a much more satisfactory result (eq 4), but we encountered much difficulty in removing the SEM group at a later stage; the SEM group was thus deemed to be unsuitable for the present purpose.”
Lesson: Higher yield is not always better! Sometimes, we have to accept lower yields in the early steps of a synthesis to finish it!
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Examples of SEM PROTECTION in Organic Synthesis
Our first example shows the orthogonality of SEM and MOM [3]. Harsher conditions are needed to remove SEM, but our MOM groups survive at 100 °C without issue.
The second example is really interesting.
Question: Although this reaction uses similar conditions as our first example, we are far from our nice 98% yield of a single product. Can you guess the structures of the three products?
Solution: OK, first of all we do see a MOM group but the first example taught us that these do not fly off when exposed to fluoride sources (unless we cook them in e.g., HF).The first insight is that the triethylsilyl (TES) group might also be a victim of the fluoride deprotection. So we might expect a mixture of SEM- and TES-deprotected products.
Indeed, product 1 is SEM- and TES-deprotected one whereas product 2 is only TES-deprotected.
But what about product 3?
Beware of interrupted deprotections!
Instead of beta-elimination and release of ethylene (and formaldehyde), it looks like we have a proto-desilylation. The ethyl group remains on the ether, and we basically have a one-carbon extended MOM group now. Why could this SEM group just not be bothered to leave? Uhm, we do not know. That’s chemistry for you.
We have seen a few of these tricky questions with other protecting groups, where, e.g., the presence of an intramolecular nucleophile can lead to side products.
As we mention often, many alcohol protecting groups can also be used to protect carboxylic acids or amines. As we see in our third example [5], SEM can also used to protect the nucleophilic nitrogen in heterocycles like imidazole.
This acidic deprotection was particularly sluggish. At 25 °C, >200 equivalents of TFA were added over two batches and ultimately just gave 20% yield. As we see, SEM is indeed hard to remove!
We’re done! If you learned something, make sure to check out my other articles on protecting groups, my page or my videos!
SEM Protection experimental procedure [6]
“To a dry 100 mL round-bottom flask under argon was added anhydrous DMF (40 mL) and NaH (0.293 g, 60%, 7.31 mmol); then the solution was cooled to 0 °C. In a separate flask the alcohol (1.021 g, 4.87 mmol) was dissolved in DMF (10 mL), and then this solution was added dropwise by cannula to the NaH/DMF mixture. The reaction mixture was stirred at 0 °C for 2 h, and then 2-(trimethylsilyl)ethoxymethyl chloride (1.067 g, 6.33 mmol) was added. After 10 h saturated NH4Cl solution (10 mL) was added, and this mixture was extracted with ethyl acetate. The combined organic layers were washed with water (3 × 25 mL) and brine (1 × 25 mL), dried over Na2SO4, and then concentrated to give a crude solid which was purified using flash chromatography (hexanes–ethyl acetate, 1:1) to give 12 as a light brown solid (1.25 g, 78%).”
SEM deprotection experimental procedure [6]
“To a 100 mL round-bottom flask, the alcohol was added (0.396 g, 0.84 mmol) and dissolved in DMF (50 mL). To this was added tetramethylethylenediamine (0.293 g, 2.53 mmol) and TBAF (1.0 M in THF, 2.53 mL, 2.53 mmol), attached a reflux condenser and set the reaction for heating at 45 °C for 20 h. After confirming the completion of the reaction by LCMS (m/z 340), the reaction was allowed to cool to room temperature and to it was added saturated solution of NH4Cl. The contents were transferred to a separatory funnel containing 100 mL of water. Extraction was done using ethyl acetate, and the combined organic layer was washed with water, followed by brine, dried over sodium sulfate, and evaporated the solvents to give a crude solid which after flash chromatography purification using hexane–ethyl acetate (1:1) gave 8 as a buff solid (0.202 g, 71%).”
SEM Protecting Group References
[1] β-(Trimethylsilyl)ethoxymethyl chloride. A new reagent for the protection of the hydroxyl group | Bruce H. Lipshutz, Joseph J. Pegram | Tetrahedron Letters 1980, 21, 3343
[2] P. J. Kocienski: Protecting Groups (Thieme)
[3] P. G. M. Wuts, T. W. Greene: Greene’s Protective in Organic Synthesis (Wiley)
[5] Discovery of Bis-imidazolecarboxamide Derivatives as Novel, Potent, and Selective TNIK Inhibitors for the Treatment of Idiopathic Pulmonary Fibrosis | Vladimir Aladinskiy, Chris Kruse, Luoheng Qin, Eugene Babin, Yaya Fan, Georgiy Andreev, Heng Zhao, Yanyun Fu, Man Zhang, Yan Ivanenkov, Alex Aliper, Alex Zhavoronkov, Feng Ren | J. Med. Chem. 2024, 67, 19121
[6] Tale of Two Protecting Groups—Boc vs SEM—for Directed Lithiation and C–C Bond Formation on a Pyrrolopyridazinone Core | Reji N. Nair, Thomas D. Bannister | Org. Process Res. Dev. 2016, 20, 1370
The MEM protecting group protects alcohols as stable acetals. MEM is removed with protic acids(H+) and more importantly, Lewis acids.
2-Methoxyethoxymethyl (MEM) is an acetal-type protecting group for alcohols. 🫡 Here’s what you’ll learn here:
Table of contents
👀 MEM is related to the simpler MOM protecting group. It’s rather oldschool, but the additional ether group brings an interesting twist!
What is the MEM Protecting Group?
The MEM group belongs to the class of acetal (‘double-ether’) protecting groups which are much less reactive than their free alcohol counterparts. Fun fact: The MEM group was introduced in 1976 by the legendary chemist E. J. Corey and co-workers [1], just like the TBS/TBDMS and allyl protecting groups.
MEM is stable under various conditions, including strong bases, reductants, organometallic reagents, oxidants and mild acids. What makes it unique is the selective cleavage with Lewis acids (see below).
MEM Protection Mechanism
The protection conditions are identical to MOM: 1) Treatment with MEM chloride and DIPEA (N, N-diisopropylethylamine) or another weak base. Deprotonation occurs after nucleophilic attack. 2) Treatment with a strong base like NaH and MEM chloride. Experimentally / in the lab, only base is added to the alcohol first – and only after some time (e.g., 1h to ensure the alkoxide is formed), MEMCl is added.
MEMCl is another case of an activated alkylating agent. The adjacent oxygen can facilitate departure of the chloride, creating a highly electrophilic oxonium ion. (No surprise it’s also bad for you – it alkylates your DNA!)
MEM deprotection mechanism 1: Bronsted Acid (H+)
Just like MOM or THP, we can get rid of MEM with acid, tough typically stronger / more forcing conditions are required. Thus, it is possible to selectively remove THP, MOM or PMB groups in the presence of MEM. The simplest mechanism starts with protonation of our protected oxygen in the acetal system. This cation can release of our free alcohol and form some type of byproduct (depending on solvent).
The alternative / indirect mechanismwould be protonation of the other oxygen atom, going through a hemiacetal intermediate. With water in the solvent or upon work-up, this unstable species releases formaldehyde and gives our free alcohol.
So what makes MEM unique versus MOM? Due to the presence of an additional ether oxygen, the acetal is much more labile when exposed to Lewis acids. Due to this additional lone pair, we have bidentate coordination to metals such as i) ZnBr2, ii)TiCl4, – or like we’ve seen for MOM, also more Lewis acidic species like iii)TMSI.
The productive mechanism is now kind of the opposite as before – it is the oxygen of our protected alcohol that cleaves the protecting group. This oxonium intermediate is ultimately hydrolysed (either water present in the reaction or upon work-up), releasing formaldehyde and giving our deprotected product.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Examples of MEM in Organic Synthesis
Our first example [2] is really chill. Simple introduction of MEM, just like we’ve talked about.
Too easy for you? Well, check this one out:
Upon treatment of the compound above, two products were observed in significant yield. What are they?
One of the products surely is the normal MEM-deprotected alcohol. After all, this is an article about MEM and we’ve just seen than ZnBr2 will act as a Lewis acid and remove the group. We cannot say with certainty, but this is probably the major product 1.
But what about product 2? If you have no ideas, go back to the mechanism above and look at what happens after initial break-down of MEM!
Always watch out for neighbouring group participation!
The answer is very cool: Our starting material has a sneaky hydroxyl group that can attack our oxonium intermediate. This occurs because the intramolecular reaction is very fast – even despite the tertiary alcohol being very hindered. This means that we do not go through the intermolecular bromide-hydrolysis pathway, but rather form a cyclic acetal!
That’s it! If you learned something, make sure to check out my other articles on protecting groups, my page or my videos!
MEM Protection experimental procedure [2]
“To a solution of alcohol (4g, 40 mmol) in CH2Cl2 (100 mL)under N2 was added MEM chloride (7.5g, 60mmol) and DIPEA (7.8g, 60mmol) at 25 °C and the reaction mixture was stirred at rt for 5 h. Water (30 mL) was added to the reaction mixture and CH2Cl2 was used to extract the mixture. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to give 7.4g of crude product. Flash chromatography of the residue on silica gel gave 6.1g (80%) of MEM protected alcohol as a colorless liquid.”
MEM Protecting Group References
General: P. G. M. Wuts, T. W. Greene: Greene’s Protective in Organic Synthesis (Wiley)
[1] A new general method for protection of the hydroxyl function | E.J. Corey, Jean-Louis Gras, Peter Ulrich | Tetrahedron Letters 1976, 17, 809
[2] Total Synthesis of (±)-Deoxypenostatin A. Approaches to the Syntheses of Penostatins A and B | Barry B. Snider, Tao Liu | J. Org. Chem. 2000, 65, 8490
The MOM protecting group protects alcohols as acetals. MOM is introduced with MOMCl and removed with acid (H+).
Methoxymethyl is the simplest acetal protecting groupfor alcohols. 🫡 Here’s what you’ll learn:
Table of contents
👀 MOM is commonly used, and is similar to PGs like THP, so it’s a “must-know” for students!
What is the MOM Protecting Group?
Protecting groups serve to minimize unwanted side reactions during organic synthesis. When protecting alcohols, methoxymethyl ethers actually form a type of acetal (‘double-ether’). These are much less reactive than the free alcohol.
Remember – just like with other protecting groups – that different nucleophiles can be protected with the same protecting group. This is why MOM is also used to protect phenols, carboxylic acids (still a nucleophilic oxygen) as well as amines (nucleophilic nitrogen). Introduction and removal (typically acid) follow the same logic, so most study materials focus on just the protection of alcohols.
MOM Protection Mechanism
Two protection conditions are most common for alcohols: 1) Treatment with MOM chloride and DIPEA (N, N-diisopropylethylamine) or another weak base. Here, deprotonation occurs after nucleophilic attack. 2) Treatment with the strong base NaH (or KH) and MOM chloride. Here, deprotonation occurs first and nucleophilic attack is second. Experimentally / in the lab, only base is added to the alcohol first – and only after some time (e.g., 1h to ensure the alkoxide is formed), MOMCl is added.
Note that the lone pairs on the oxygen on MOMCl actually activate the departure of the chloride. This creates a highly reactive, electrophilic oxonium ion which is captured by the alcohol. This makes MOMCl a very powerful alkylating agent and carcinogenic (it alkylates your DNA base pairs which is not what you want). So, special care in the lab is required.
A safer alternative protocol is the following: 3) Use of dimethoxy methane and an acid. This reaction is different as it is an acetal exchange reaction and uses an excess of reagent to drive the equilibrium.
mOm deprotection mechanism
The standard MOM deprotection is acidic hydrolysis. Protonation activates the acetal system towards release of our free alcohol. Less importantly, the remaining stabilized cation forms a byproduct after trapping by the solvent. This is pretty similar to the THP deprotection.
Note that you can also draw the alternative / indirect mechanism with protonation at the other oxygen, leading to a hemiacetal intermediate and ultimately our free alcohol upon elimination of formaldehyde.
Exemplary deprotection conditions are: i)HCl in aqueous EtOH; ii) TFA (trifluoroacetic acid) in dichloromethane; iii) PPTS (pyridinium p-toluenesulfonate) in tBuOH.
Alternatively, reactive electrophiles / Lewis acids like TMS+ can also be used to remove MOM groups. A key method is use of TMSBr (the mechanism follows a similar logic of nucleophilic attack).
Classes often teach you a single method (to preserve your sanity) of protection or deprotection, but having different options at our disposal helps in the lab when we deal with complex molecules that have other protecting groups!
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Examples of MOM in Organic Synthesis
Now that we now the basics, let’s check out two use cases of MOM and connect it to other protecting groups we have learned about.
The first example [1] shows that lability does not equal lability. We’ve learned that the PMB protecting group can be cleaved under acidic conditions. However, HCl (generated in situ from AcCl and MeOH) here selectively removed the MOM group while retaining the PMB group. Chemistry is pretty experimental!
The second example [2] is a fancy regioselective introduction of the MOM group. This procedure achieves MOM protection at the more sterically hindered alcohol of a vicine diol which should be less reactive with reagents like MOM-Cl (secondary vs. primary alcohol). The trick here is to create the orthoester first as a detour. It can then be reduced with DIBAL-H which exhibits preference in coordination and hydride reduction, leaving the MOM group hanging at the more hindered alcohol!
Appreciate you reading the full article! Feel free to check out other protecting groups, my page or my videos!
MOM Protection experimental procedure [3]
“An oven-dried 3-neck 500 mL round-bottom flask equipped with a stir bar was charged with alcohol 16 (15.5 g, 78.18 mmol, 1.0 eq.), DIPEA (40.41 g, 312.72 mmol, 4.0 eq.) and DCM (160 mL) under Ar. The resulting suspension was cooled down to 0 °C and freshly distilled MOMCl (18.88 g, 234.50 mmol, 3.0 eq.) was added dropwise over a period of 10 min. NaI (5.80 g, 39.09 mmol, 0.5 eq) was added to the reaction solution, and the resulting mixture was allowed to warm to 25 °C, and stirred for 16 h. After completion of the reaction, the reaction mixture was quenched with saturated ammonium chloride solution (300 mL) and diluted with DCM (100 mL).The two layers were separated and aqueous layer was extracted with DCM (2 × 100 mL). Combined organic phases were washed with saturated sodium chloride solution (1 × 100 mL),dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography to give 17 (17.5 g, 92% yield) as colorless liquid.”
MOM deprotection experimental procedure [3]
“31 (68 mg, 0.142 mmol, 1.0 eq.) was dissolved in DCM/TFA = 15/1 (3 mL) at 25 °C. The resulting suspension was stirred at 25 °C for 12 h, when TLC analysis of the crude mixture showed full conversion. The reaction mixture was diluted with DCM (2 mL) and treated with sat. aq. NaHCO3 (4 mL). The layers were separated and the aqueous phase was extracted with DCM (2 x 3 mL). Combined organic phases were washed with sat. aq. NaCl (1 × 5 mL),dried over anhydrous MgSO4 and concentrated under reduced pressure to give the crude product. The crude residue was purified by preparative TLC to provide isorosthin L (35 mg, 71% yield) as a white solid.”
mom Protecting Group References
[1] P. G. M. Wuts, T. W. Greene: Greene’s Protective in Organic Synthesis (Wiley)
[2] A convenient procedure for the regioselective monoprotection of 1,n-diols | M. Takasu, Y. Naruse, H. Yamamoto | Tetrahedron Lett. 1988, 29, 194
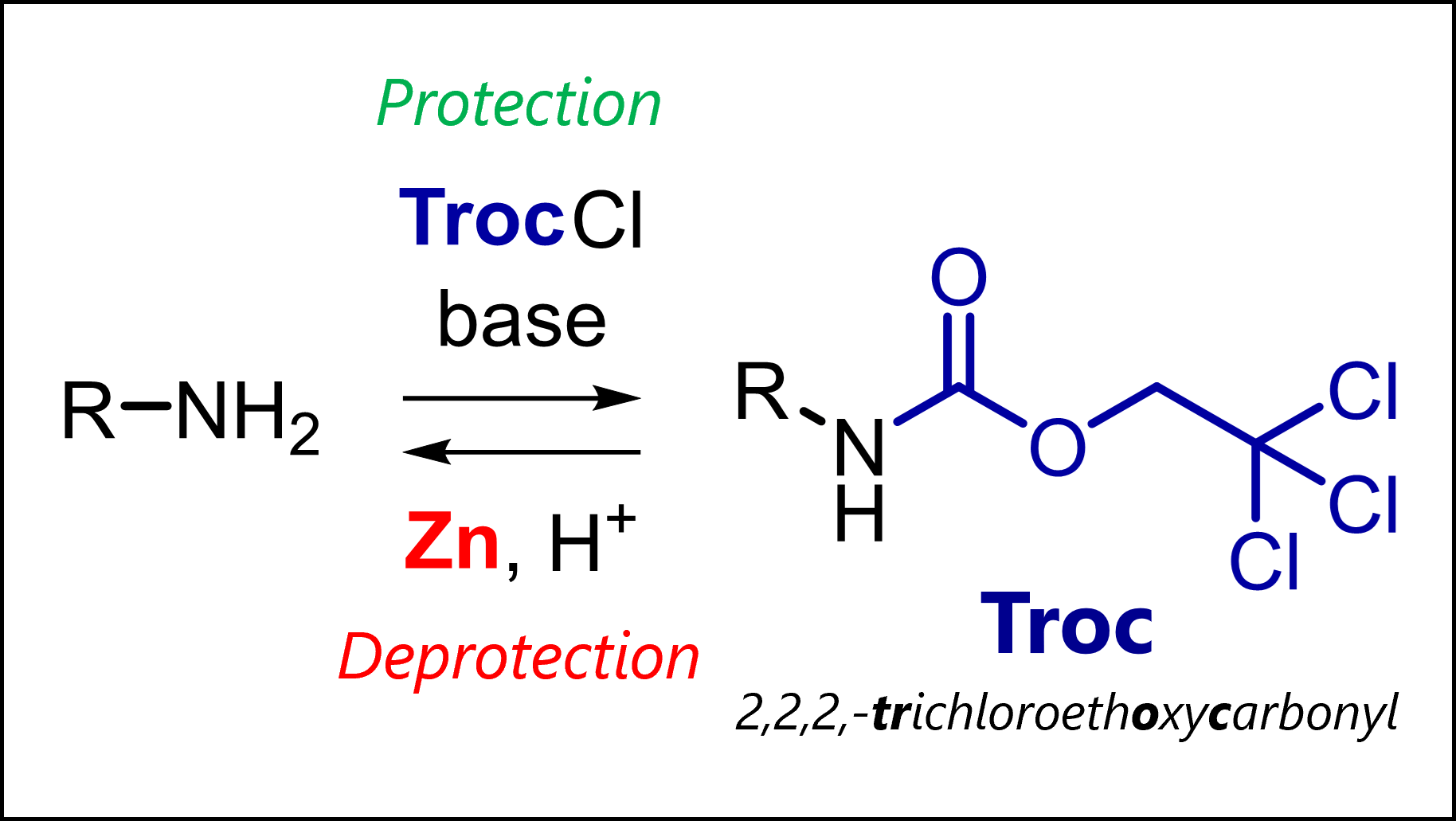
The Troc protecting group protects amines in organic synthesis. Troc is introduced with TrocCl and deprotected by reduction (Zn).
🫡 Hello! Here’s what you’ll learn here:
Table of contents
👀 Left: 3D model of Troc (chlorines in green!)
What is the Troc Protecting Group?
Troc (2,2,2-trichloroethoxycarbonyl) can convert amines or alcohols into stable carbamate or carbonate derivatives. This leads to a similar effect that we see in groups like Fmoc or Boc: the previously nucleophilic amine or alcohols loses its reactivity (driven by delocalization of electrons into the carbonyl system).
The nice thing is that Troc is orthogonal as the deprotection conditions for Fmoc (base), Boc (acid) or silyl groups like TBS (fluoride) do not remove it. Instead, it has a unique mechanism of reductive beta-elimination to release the free group.
Did you know: The Troc group was introduced by the legendary Robert Burns Woodward in the 1960s? (see below)
Troc Protection Mechanism
The Troc protecting group is introduced by reacting the free amine or alcohol with 2,2,2-trichloroethyl chloroformate (Troc-Cl) with addition of a base.
Most common protection conditions are Troc-Cl with pyridine in CH2Cl2 or THF. If the starting material is very polar, Troc-Cl with NaOH or NaHCO3in water.
Troc Deprotection Mechanism
The Troc protecting group can be removed with certain reductive methods which all function via beta-elimination. Most common is use of zinc or other single-electron reductants that reduce the terminal carbon. The beta-elimination gives a free carbamate – an intermediate seen in mechanisms of many other protecting groups – that rapidly decarboxylates to the deprotected amine (or alcohol).
Most common deprotection conditions are Zn powder in THF/H2O or so-called couples/alloys consisting of mixtures of Zn-Pb or Cd-Pb. More rarely used, reduction with electrolysis also removes the Troc group.
Always remember protecting group stability or lability are always general (e.g., here: removal by reduction). Nothing in chemistry (or life) is black and white 🙂 Here is a neutral method (not reductive) using trimethyltin hydroxide. [1]
A fascinating point is that under the right conditions, Me3SnOH actually deprotects Troc selectively in the presence of methyl esters (you would expect that the methyl ester is quite labile to hydrolysis with hydroxide).
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Troc protecting group in total synthesis
The very first introduction of the Troc protecting group was already… very advanced! Woodward and co-workers used the starting material below in the synthesis of cephalosporin C, an antibiotic natural product. [2]
What is the product after treating this molecule with Zn in aqueous AcOH?
This is a cool example where just one of the three groups decarboxylates. The others are not true Troc groups but were instead masking the free acid. If we do not have a free carbamate or carbonate after reductive elimination, we do not see a decarboxylation.
Below you can find typical Troc protection and deprotection conditions.
Troc Protection conditions [3]
To a solution of the alcohol (0.80 g, 1.48 mmol) in methylene chloride (30 ml) at 0 °C was added pyridine (0.96 ml, 11.84 mmol, 8 equiv) followed by 2,2,2-trichloroethyl chloroformate (0.8 ml, 5.92 mmol, 4 equiv), and the reaction mixture was stirred at 0 °C for 1 h. Saturated aqueous sodium bicarbonate (50 ml) was added and the organic layer was separated. The aqueous layer was extracted with methylene chloride (3 x 50 ml), and the combined organic layers were dried over anhydrous sodium sulfate and concentrated in vacuo. Purification by flash column chromatography (2% EtOAc/hexanes) afforded protected product (0.98 g, 93%) as a colorless oil.
Troc deprotection conditions [4]
To a solution of protected Troc-amine (40 mg, 57 µmol) in 4 mL of MeOH was added activated zinc (400 mg). The mixture was stirred at 25 °C for 5 min, and glacial HOAc (4 mL) was added. The mixture was heated at 60 °C for 30 min, cooled and concentrated under reduced pressure. The residue was treated with 5 mL of 5% aqueous NaOH, and the solution was extracted with EtOAc (5 × 5 mL). The combined extracts were washed with brine, dried over anhydrous K2CO3, and concentrated under reduced pressure. Flash chromatography on silica gel (100:1 CH2Cl2/MeOH) gave 25 mg (86%) of the free amine as a viscous oil.
TROC Protecting Group References
P. G. M. Wuts, T. W. Greene: Greene’s Protective in Organic Synthesis (Wiley)
[1] Highly Chemoselective Deprotection of the 2,2,2 Trichloroethoxycarbonyl (Troc) Protecting Group | Barry M. Trost, Christopher A. Kalnmals, Jacob S. Tracy, and Wen-Ju Bai | Org. Lett. 2018, 20, 8043−8046
[2] The Total Synthesis of Cephalosporin C. | Woodward, R. B.; Heusler, K.; Gosteli, J.; Naegeli, P.; Oppolzer, W.; Ramage, R.; Ranganathan, S.; Vorbruggen, H. | J. Am. Chem. Soc. 1966, 88, 852− 853
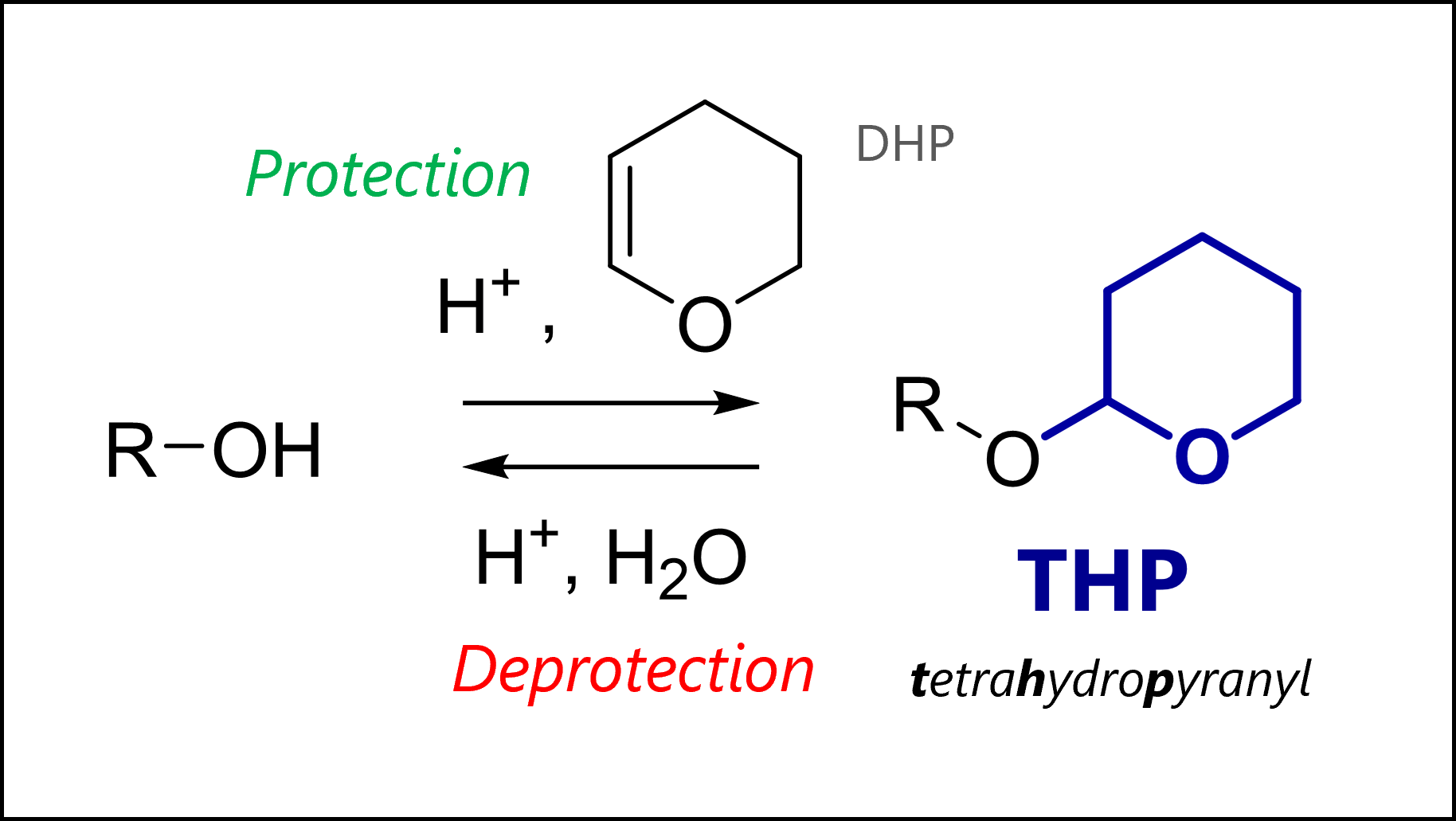
Tetrahydropyranyl ethers were one of the first protecting groups for alcohols. Nowadays, they are seen less commonly, though still used. The THP group is easily removed under acidic conditions (mechanism below) and stable to organometallic nucleophiles, electrophiles (as the protected oxygen is less nucleophilic), reduction or base. The protected THP ethers are actually a type of acetal (‘double-ether’).
THP Protection Mechanism
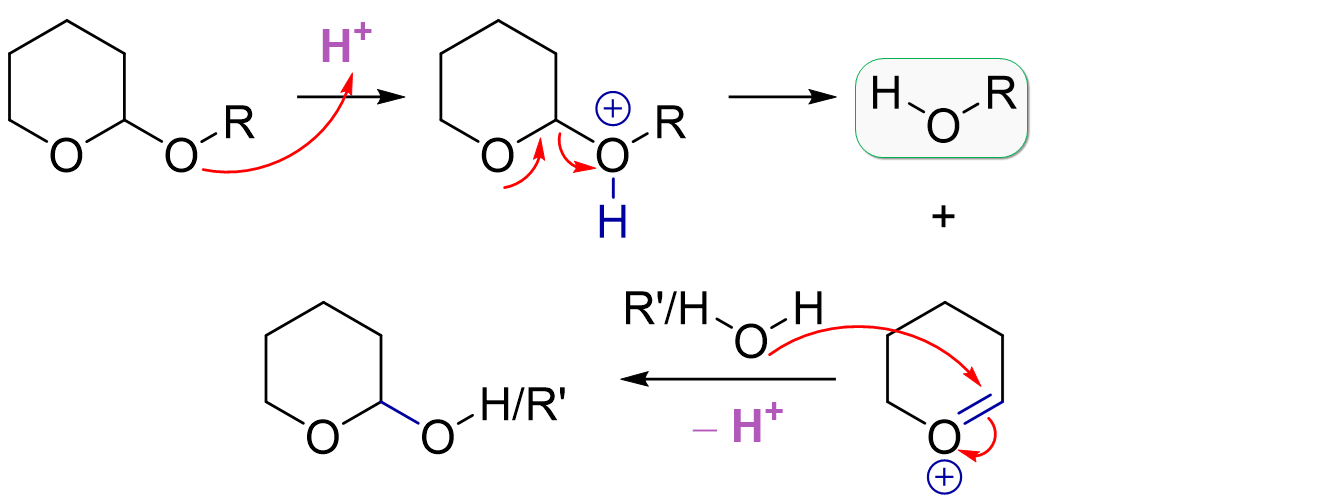
THP protection uses acid catalysis and 3,4-dihydro-2H-pyran or DHP. The mechanism proceeds by THP pre-activation with acid, leading to a stabilized cation. Here, the oxonium is drawn but you can imagine the other resonance form with the positive charge on the carbon which is ultimately where the ion is most electrophilic. Our free hydroxyl group then attacks the carbon in a nucleophilic addition, and loses a proton to give the protected THP ether. The last step regenerates our acid catalyst.
The most common protection conditions are catalytic TsOH or pyridinium p-toluenesulfonate (PPTS, a form of TsOH with lower acidity) together with 3,4-dihydro-2H-pyran in dichloromethane.
THP Deprotection Mechanism
THP deprotection is similar to MOM deprotection and actually also similar to THP protection. Acid catalysis activates the acetal system towards dissociation of our initially protonated alcohol. Again, it’s the same stabilized cation intermediate but based on the choice of solvent used, we have different potential byproducts. The solvent is obviously present in large excess, so it will preferentially attack the carbocation instead of our just liberated hydroxyl group. For example, methanol gives the methyl-substituted THP ether while use of water would give the free hydroxyl group (this can open to the linear aldehyde).
The most common deprotection conditions are AcOH:THF:H2O or PPTS in EtOH.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
THP protecting group diastereomers
One of the drawbacks of the THP protecting group versus the TBS protecting group, beyond its lower stability, is that it introduces a second chiral center. If our starting material has already at least one chiral carbon, we form diastereomers. This can complicate the separation and identification (e.g., NMR) of products – because as you know, diastereomers have different physicochemical properties.
Interestingly, some older research [1] tried to make use of this ‘drawback’. In this work, the chemists used a THP-derivative as a chiral auxiliary for nucleophilic additions to an aldehyde in the molecule.
In these derivatives, one side of the aldehyde is shielded from nucleophilic attack while the other is exposed. This leads to very high diastereoselectivity at the newly formed carbon (a tertiary alcohol). It’s not terrible useful but interesting that a protecting group can be used to exert diastereoselectivity. You could imagine this potentially being useful in some complicated total syntheses.
“To a 100-mL, single-necked, round-bottomed flask equipped with a magnetic stir-bar, argon inlet with septum, was charged with dihydropyran (1.5 equiv), followed by CH2Cl2 (15 mL) and PPTS (177.4 mg, 0.706 mmol, 0.1 equiv). The contents were cooled to 0 °C in an ice-bath. Then a suspension of iodobenzyl alcohol 9 (2.08 g, 7.06 mmol) in CH2Cl2 (10 mL) was added at 0 °C over 10 min. After addition, the contents were warmed to rt. Dihydropyran (0.97 mL, 10.59 mmol, 1.5 equiv) was added again to the mixture after 30 min, because the starting material was still observed by TLC analysis. After another 30 min of stirring, H2O (50 mL) was added and the mixture was extracted with CH2Cl2 (3 x 50 mL). The organic layers were combined and washed with brine (2 x 50 mL), dried with Na2SO4, filtered and the solvent was removed under reduced pressure by rotary evaporation. The crude material was further purified using column chromatography (SiO2, 70 g; hexanes/EtOAc, 3:1) to afford (2.67 g, >99%) THP ether 26 as a colorless wax.”
THP deprotection experimental procedure [3]
“To a solution of alkene 12 (38.6 mg, 0.047 mmol) in 2-propanol (0.95 mL), p-toluenesulfonic acid monohydrate (21.7 mg, 0.114 mmol) was added at 0 °C and stirred for 17 h at room temperature. The reaction mixture was diluted with water, extracted with dichloromethane, washed with brine, and dried over sodium sulfate. The residue was purified by thin layer chromatography (hexane/ethyl acetate = 5/1). Alcohol 13 (34.6 mg, quant.) was obtained as a colorless oil.”
THP Protecting Group References
P. G. M. Wuts, T. W. Greene: Greene’s Protective in Organic Synthesis (Wiley)
[1] The tetrahydropyranyl group as a chiral auxiliary for the nucleophilic addition to α-alkoxy ketones | André B. Charette , Abdel F. Benslimane , Christophe Mellon | Tetrahedron Letters 1995, 36, 8557
[2] Total synthesis of (+)-papulacandin D | Scott E. Denmark, Tetsuya Kobayashi, Christopher S. Regens | Tetrahedron 2010, 66, 4745
[3] Total Synthesis of Eutyscoparol A and Violaceoid C | Takatsugu Murata, Takuto Iwayama, Teppei Kuboki, Shotaro Taguchi, Shou Tsugawa, Takumi Yoshida, Hisazumi Tsutsui, Ayana Shimauchi, Yukiho Kosaka, Isamu Shiina | Asian Journal of Organic Chemistry 2024, 13, e202400148
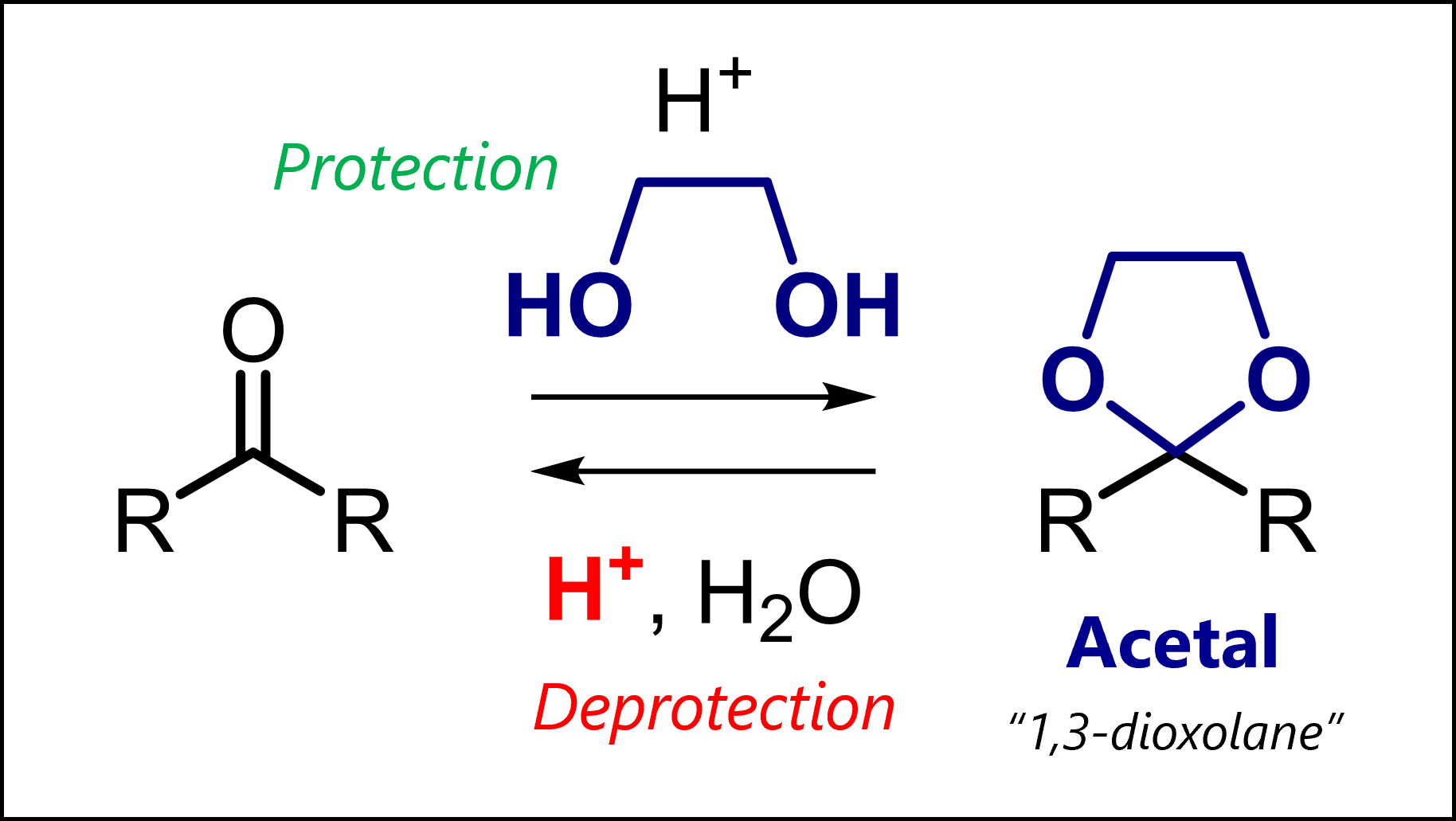
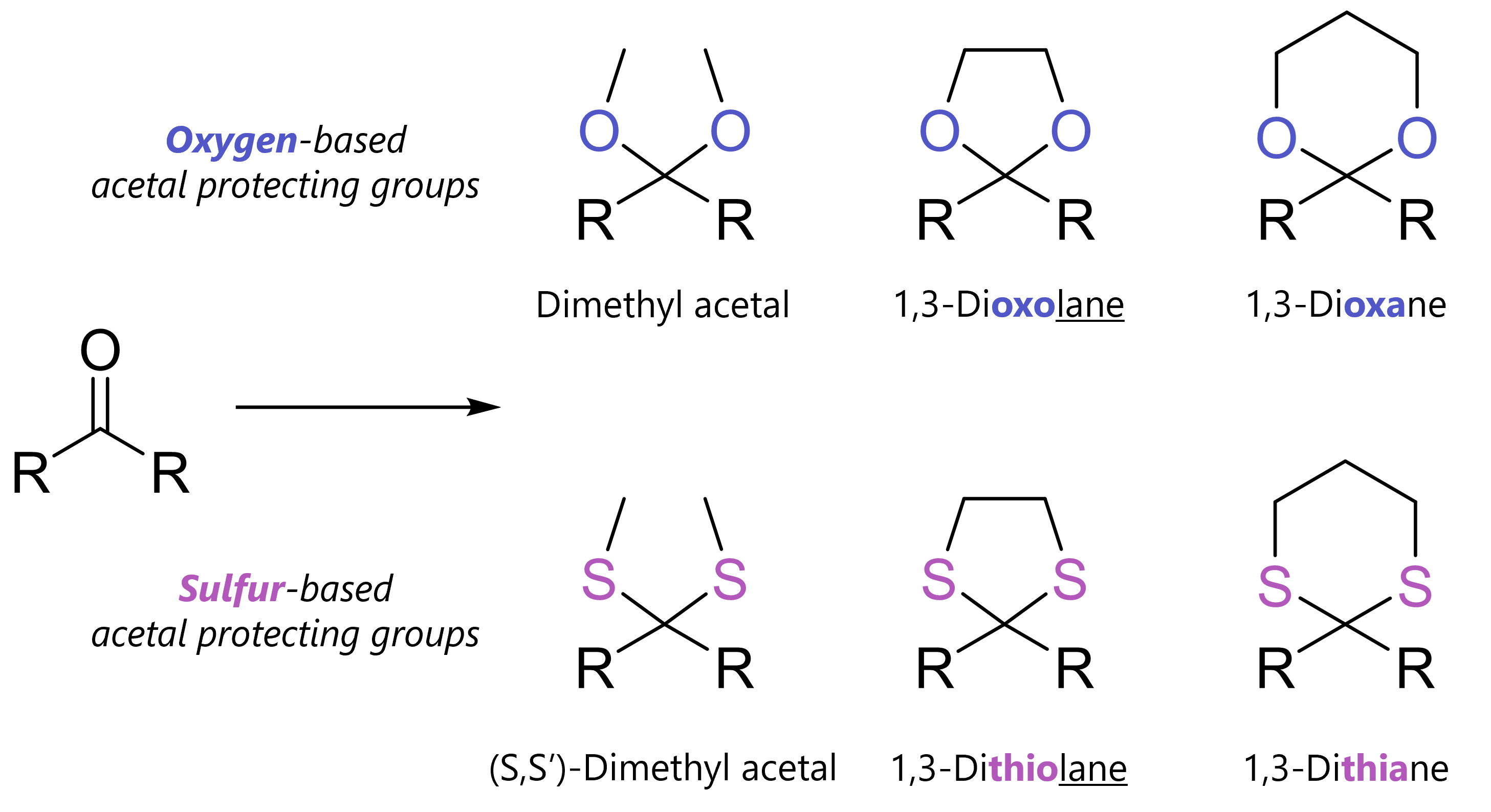
Acetal protecting groups protect carbonyls from bases, nucleophiles and hydride reduction. Among many variants, most common are dimethyl acetals, 1,3-dioxolanes and 1,3-dioxanes.
Table of contents
There is no single acetal protecting group! Rather, this is a broader family of similar protecting groups.
➡️ This article explains properties and mechanisms of acetals (protection and deprotection).
What Are Acetal Protecting Groups?
Acetals always protect carbonyl compounds. But how? This is where the variety can come from. On one hand, acyclic acetals form by reaction with an alcohol (-OH) or thiol (-SH) and catalytic acid. On the other, cyclic acetals form when carbonyls react with a diol or dithiol and catalytic acid.
Do you already know if cyclic or acyclic acetals are more stable? Why? (see below)
You will know that carbonyls are nucleophilic at the carbon. Any acetal protecting group renders it stable to these nucleophiles: aqueous and non-aqueous bases, organometallic reagents and hydrides. As always, we want to avoid unwanted reactions of one group (here, the carbonyl) to instead perform chemistry at another functional group. As we will see below, formation of acetals involves a two-step mechanism, including nucleophilic attack and subsequent dehydration, which drives the equilibrium towards product formation.
Difference between Acetal and Ketal
You might not be aware, but back in the day, people used to separate acetals – made from aldehydes – and ketals – made from ketones. Nowadays, acetal is the umbrella term that describes both – while ketal remains restricted to ketones (link to IUPAC definition).
Types Of Acetal Protecting groups
As mentioned, there are several relatives in the acetal protecting group family. The good thing is that they work very similarly!
=> You should simply know that acetals can be oxygen-based or less commonly, sulfur-based. The simplest acyclic acetal is the dimethyl acetal. Cyclic acetals have five-membered rings (1,3-dixolane; 1,3-dithiolane) six-membered rings (1,3-dioxane, 1,3-dithiane).
Acetal protection mechanism
As an example, 1,3-dioxolanes are prepared by treating carbonyls with ethylene glycol and acid. => Acetal protection or acetalization requires catalytic acid to activate the carbonyl (but only catalytic because the proton is regenerated in the final step) => Acetalization is a condensation as the original oxygen is kicked out as water
Typical conditions: Ethylene glycol and cat. TsOH (acid) in C6H6 as solvent at reflux.
Because every reaction is an equilibrium (imagine the arrows also going from right to left), chemists use ways to remove water from the reaction to ensure it can’t react back. For acetalizations, this involves using a Dean-Stark trap. The Dean-Stark trap is a glassware that collects water formed in a reaction through an azeotropic distillation. You might have heard about it – if not, does not matter. This is a physical removal – alternatively, dehydrating agents like trimethyl orthoformate can chemically remove the water by reacting with it (“scavenger”).
The same mechanism applies if we use other diols (e.g., to form six-membered 1,3-dioxanes), alcohols (e.g., methanol to form dimethyl acetals) or thiols to form sulfur-based acetal protecting groups.
Acetal Deprotection Mechanism
Deprotecting acetals is very similar to introducing them! The most common is an acid-catalyzed hydrolysis. Again, make sure you understand why it only requires catalytic acid (i.e., less than 1 “equivalent” of moles).
Typical conditions: Cat. pyridinium tosylate PPTS or HCl (as the acid) in a mixture of water (for the deprotection) and an organic solvent (to dissolve the starting material).
The sulfur-based acetals are special as they can also be removed with heavy metal salts – so Lewis acids like mercury(II) or silver(I) – or oxidants. The oxygen-based acetals are stable to these conditions. We will go into 1,3-dithianes and 1,3-dithiolanes into more detail in another post.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Acetal protecting group stability
Are cyclic or acyclic acetals more stable?
Cyclic acetals are more stable than acyclic ones. Why? Acidic hydrolysis starts with protonation (catalytic acid), and goes via the oxonium intermediate.
For the cyclic acetal, the newly released hydroxyl group is still in the same molecule – so the reverse reaction would be an intramolecular reaction which is very fast (entropically favored).
For acyclic acetals, formation of the oxonium cleaves off an alcohol as a separate molecule. Because the deprotection is in aqueous solvent, we have a lot of water molecules around. It is now much more likely water will attack the oxonium (leading to deprotection of the carbonyl) instead of the alcohol attacking. This is because we only have 1 molecule of alcohol formed for 1 molecule of starting material; on the other hand, we have a large excess of water molecules.
“Why do they call it organic chemistry? Nothing about this feels natural.” A student, somewhere out there
Organic chemistry is often perceived as one of the most challenging subjects for students. Unfortunately, this creates a lot of anxiety and misconceptions about its complexity and requirements, particularly for students in adjacent fields like medicine, pharmacy or biology.
But why the bad reputation?
Concepts: Yes, organic chemistry is complex! After all, it deals with the literal wizardry of transforming living matter. The complexity (and bad teaching) can make many students feel overwhelmed – amplified by cumulative learning: missing foundational concepts makes it difficult to understand advanced ones.
Representation: Organic chemistry requires abstract thinking and relies on molecular drawings that are often not intuitive or tangible. After all, we are drawing 3D things on 2D paper. Fischer projections anyone?! (I hated these) (how to change: look at 3D model, simulate it online, practice practice)
Process:Teachers rush through content without explaining it properly, leading students to fall behind completely and/or rely on memorization to stay afloat (how to change: try to maximize the support at your uni with open doors, ask more senior students – if no support environment, look online and try to understand the why, write as many things out as you can to build your mental muscle…)
But do not despair – organic chemistry can be more accessible than you might think. Here are some ways to help you address these issues.
These tips are not some crazy 200 IQ approaches, but they require putting in the work – thoughtfully. It’s like getting fit: it’s relatively obvious what you need to do (eat healthy, work out, sleep…), but the challenging part is doing it!
1. Organic Chemistry Concepts
Build the foundation: Whatever level of class you are taking, first go back to your older materials. For introductory organic chemistry lectures this means understanding basic models like the atomic model (electrons, protons, neutrons) and orbitals and hybridization (if relevant in your class). Why do molecules inherently repel each other, and why do reactions occur nevertheless? What is a chemical reaction? Then, revisit concepts like electronegativity, nucleophilicity and electrophilicity (link), … I might work on a full list of concepts in future.
Understand the functional groups(link): Learn the structures of the most common functional groups in organic chemistry. Sorry, but there is some memorization involved. It’s just like learning a new language: you need to memorize at least new words! For this, you have to understand what a skeletal formula is, and how it differs to normal molecular drawings (next section). Again, I will go into more details in future but here are two easy exercises: =>Get a list of the 15-20 most common functional groups and draw their structures in skeletal formulas. Can’t remember a particular one, like the nitro group and its partial charges? Well, simply draw it 5 times for 2-3 days. You will know all of them in no time. Then, look at them and think about their similarities: For example, a thiol group is like a hydroxyl group (or an alcohol), just that the oxygen is replaced by sulfur which is in the same “group” in the periodic system. An ester is similar to a tertiary amide, just that the -OR group bound to the central carbonyl carbon is replaced by a -NR2 group. Tertiary amides are similar to secondary and primary amides, just that the number of alkyl rests versus hydrogens bound to nitrogen differ. => Google “pharmaceuticals” and check out their structures on Google images. Pick a random pharmaceutical – what functional groups can you identify? Which groups are ones you do not know yet? Do they look similar to any which you know already?
Work your way up the complexity of reactions: 😔 The bad news : There are a literal TON of reactions in organic chemistry. 😊The good news: Thankfully, you don’t have to know all of them, and many of them share the same logic and concepts. Here you should again start easy and build up. => Before trying to understand SN1 reactions or more difficult name reactions, first review acid-base reactions and oxidations/reductions. Then, get into additions. Then, substitutions and eliminations (SN2 and E2 first because they are just one step – then, SN1 and E1). After these, you can start to look at name reactions and complex transformations. For example, if you have are discussing electrophilic aromatic substitutions in class, you need to know how much simpler additions and substitutions work!
Focus on conceptual understanding: It’s normal to memorize things at the start, but really strive to understand the “why” behind reactions and mechanisms. For example: Why does a molecule X react in a substitution – where is its nucleophilic group? Why is the reagent Y an oxidant – which atom has a high oxidation number and gets reduced? Why does the reaction occur regioselectively at this position of the aromatic ring – are there electron-donating or electron-withdrawing substituents somewhere?
Create mind maps: Look at all the reactions you have studies in your class, and try to connect them based on their similarities. Or, connect different concepts: resonance impacts many other factors like acidity of a molecule. How? Electronegativity impacts intermolecular forces like hydrogen bonds and dipole effects. How? Diastereoisomerism impacts melting and boiling points. Why
Create analogies: Whenever you find a concept confusing or can’t remember it, create an analogy. For example, nucleophiles and electrophiles can correspond to rich and broke people, respectively. Electrophiles would like to borrow the electrons of the nucleophiles, creating a strong attraction. The process of the rich lending money to the poor is the nucleophile giving its electrons to the electrophile.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
2. Representation
Master skeletal formulas: Organic chemistry relies on skeletal formulas which omit the explicit indication of carbon and most hydrogen atoms. Remember, each “bend” or end of a line represents a carbon atom, and hydrogen atoms are implied to fill the carbon’s required bonds.
Change your perspective: Molecules can be represented in various ways: Lewis structures, skeletal structures, Newman projections, Fischer projections… Each emphasizes different aspects of a molecule (e.g., stereochemistry). Practice switching between them to strengthen your spatial awareness. => Practice converting between full structures and skeletal formulas until it’s your second nature. Make sure you understand where electron pairs are present! => Convert between Newman projections and skeletal formulas. Are your Fischer projections your enemy? Well, go and solve literally 20 different problems. You will get the hang of it.
Use molecular models to visualize stereochemistry: Stereochemistry is a tricky topic because because it involves 3D thinking. Physical or digital 3D models of molecules can help you see how atoms are arranged in space. => Use online resources like MolView to visualize molecules. Struggling with chirality, enantiomers, and diastereomers? Go to town on the visualization. Rotate, inspect and understand the structures! If you have an university ChemDraw license, use Chem3D!
Practice electron pushing: Organic reactions are governed by electron movement. Curved arrows indicate this electron flow. Whenever you’re learning a new reaction, focus on understanding the arrows, which show how bonds are made and broken. If you can follow the electrons, you’ll have a better grasp on the mechanics of reactions. The same applies to resonance. Pick any molecule of your choice and draw all the different resonance structures. Why resonance structure is the most stable? How does resonance impact the reactivity of the molecule?
Color code drawings for clarity: Use different colors if that helps you. For example, always use red to indicate movement of electrons. Does the reaction involve partial charges? Color code them!
“Play chess” – think several moves ahead: At the beginning, you should write out every single step in a reaction. Once you are at the intermediate level (when you get to practicing retrosynthesis), try visualizing several transformations at once. How would my final product look like if I oxidize the alcohol in the molecule to the aldehyde and perform a Wittig reaction?
3. How to learn organic chemistry
Stay consistent with daily practice: We’ve all been guilty of cramming at some point. However, organic chemistry is best learned through regular practice. Even if you only have 15 minutes, do something related to the course every day—whether it’s reviewing notes, drawing mechanisms, or solving practice problems. The more often you engage with the material, the more familiar it becomes. => Every day, read about one new molecule of your choice. Think drugs against neurodegeneration are cool? Research the structures of Alzheimer’s drugs. What are their functional groups? How are they synthesized? Maybe you are into doing sports? Look at the different types of steroids and performance-enhancing drugs that there are! (Do NOT take them, just read about them…)
Activate your little brain cells: Look, we all are guilty of passively consuming information and entertainment. But instead of just reading textbooks or watching videos, actively engage with the content. Draw out mechanisms as you learn them, explain concepts in your own words, or quiz yourself on reaction types and mechanisms. Basically, anything you are not writing down or creating yourself, you will probably not remember!
Test yourself with practice problems: Organic chemistry is problem-solving heavy, so doing lots of practice problems is essential. Don’t be discouraged by mistakes or getting stuck – this is part of the learning process. The rate-limiting step (see what I did there?) might be how many good problems you can find – you know, ones with proper explanations. The quality of your problems will depend on your luck and teacher. I’m working on a problem book for chemistry myself – but yeah, go to town on Google and YouTube.
Emphasize understanding over memorization: I mentioned this one already – but given we are talking about the learning process here, I wanted to reiterate.
Create your own study materials: Summarize concepts, reactions, and mechanisms in your own words. Create summary sheets, mind maps (see above), or flashcards tailored to your learning style. Experiment and see what works for you! For the things that you will need to memorize (e.g., functional groups, pKa values, named reactions…), you should have a clear approach.
Team up: Organic chemistry is almost always more manageable when tackled with peers. Explain concepts to each other, discuss exercises together, and discuss tricky mechanisms. Your classmates are ahead of you? Great, you can suck out their insights and ask questions. You are ahead of your classmates? Great, help them and in doing so, you will further elevate your capabilities. Are you in university and not against earning an extra buck? Tutor students that are 1-2 years below your level.
Use office hours and ask for help early: If something doesn’t make sense, ask for help right away. Afraid of looking like a moron? Well, the odds are at least half of your class is feeling like morons as well, so don’t feel bad! Visit your professor or teaching assistant during office hours to clarify any misunderstandings. Addressing confusion early prevents small gaps in knowledge from turning into major issues later.
Use multiple resources: Don’t rely solely on one textbook or lecture notes. Different resources may explain the same concept in slightly different ways, and sometimes a different perspective makes things click. Use videos, alternative textbooks, online platforms, and forums to diversify your learning. No access to textbooks? Go to town on LibGen (google it).
Prioritize your effort: As mentioned, building the foundation is key. But once you get into intermediate territory, there are SO many things you could theoretically read about. In the best case, your textbook and/or teacher might have clear objectives or checklists of topics to know. Assess on what you need to work on most, and then go hard.
Is Organic Chemistry Hard?
So, is organic chemistry hard? Honestly – yes, it’s hard.
Is it as hard or terrifying as people make it out to be? No, not really. The great thing is that at some point, every new concept or reaction will be based on something you know already. After enough effort, there will be almost no cases where you really have absolutely no clue of how to approach a problem.
Is it the hardest subject you will study? Maybe, maybe not. Depending on your area of focus, it might come least naturally to you, versus other subjects. If you dislike chemistry and math, physical chemistry will haunt you even more.
To end on a positive: I loved organic chemistry more than any other subject but even I had been struggling with it initially. Resonance? Confusing as hell. Fischer projections? I couldn’t draw a single one correctly. The abbreviation of -Me for a methyl group (-CH3)? I thought it stood for “a generic metal” (lol). I asked A LOT of stupid “questions” (or in many cases did not ask them, which slowed down my progress). What had been a guessing game and brain-dead memorization, became almost second nature.
Whatever your journey in organic chemistry, I hope you can figure out how to make it a bit more bearable and more successful – however you define it: just passing, improving your grade, or really going beyond the class and mastering it.
I’m developing content to help students unlock and practice organic chemistry. If you’re interested, follow along!
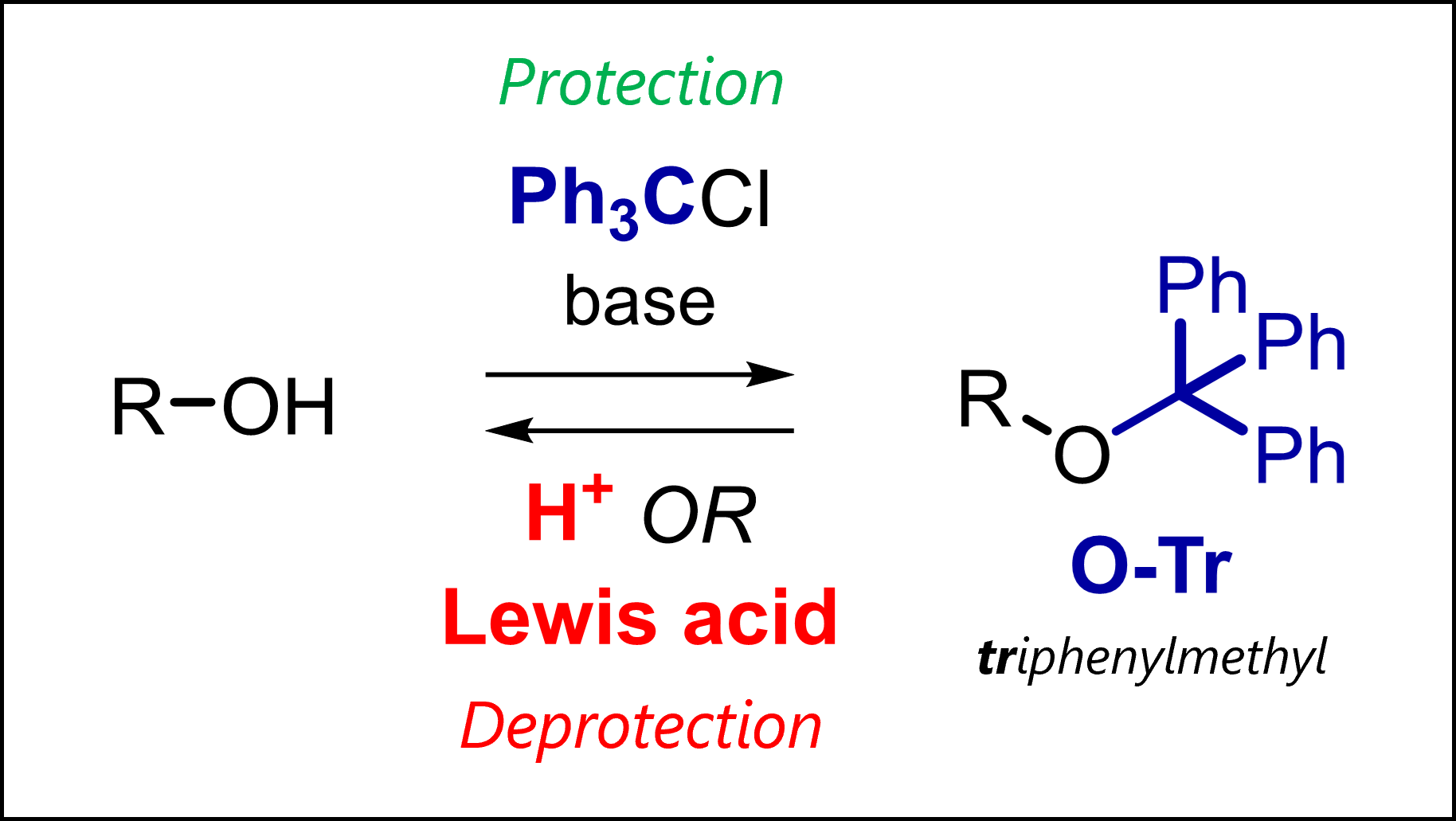
The trityl protecting group protects alcohols in organic synthesis. Tr is deprotected with acids (TFA) or Lewis acids.
The trityl group(Tr / Trt) is an acid-labile protecting group (reminds you of Boc?)! Here we cover “standard” trityl and more advanced versions.
👀 As you see in the 3D model, it’s huge!
Table of contents
What is the Trityl Protecting Group?
Trityl stands for triphenylmethyl, a group most commonly used to protect free alcohols as ethers. As seen with other PGs, amines and thiols can also be protected (as they are also nucleophilic).
Trityl ethers saw most application in carbohydrate chemistry as their hydrophobicity was useful for protection of polar building blocks. In addition, their bulky size allows for selective protection of primary hydroxyls due to sterics. Over time, silyl PGs like TBS have replaced much of the use of Tr outside of carbohydrate chemistry. Nevertheless, we can learn some things by studying it!
Trityl Protection Mechanism
Trityl protection usually uses trityl chloride in pyridine which conveniently functions as a base, capturing the HCl by-product. Added DMAP (4-dimethylaminopyridine) can function as a base but also as a catalyst. You likely remember from other protecting groups (e.g., TBS) or reactions like acylation that DMAP works as a transfer reagent via initial nucleophilic addition to the activated reagent and transfer to our group of interest (not shown above).
Watch out as there are some pages online that imply a direct SN2-like attack of the free alcohol to Tr-Cl. You should know this is impossible; quaternary carbons do not undergo SN2! Instead, the protection proceeds as a SN1 via the stable trityl cation intermediate [1]. The large size allows the selective tritylation of primary alcohols in the presence of secondary alcohols as these react much slower due to steric hindrance.
No surprise, there are other ways to introduce trityl like TrOTf (recall just like for TBS) or trityl-pyridinium tetrafluoroborate, an even more reactive transfer reagent.
Trityl Deprotection mechanism
Trityl is deprotected with Bronsted acids or less commonly also Lewis acids. In both mechanisms, the highly stable trityl cation is a common theme. This should remind you of the Boc group: The trityl cation here is basically a t-butyl cation (acidic Boc deprotection intermediate) on steroids.
Case 1: The deprotection with a Bronsted acid starts with protonation of the ether oxygen. This increases the “pull” on the O-C bond which can fragment to give our deprotected hydroxyl group. The resulting trityl cation is still reactive, so adding nucleophilic scavengers like 2-methyl-2-butene can avoid undesired reactions. By using acetic acid or formic acid, it is possible to deprotect trityl ethers in the presence of TBS ethers. If no sensitive groups are present, stronger acids like TFA obviously work as well.
Case 2: Using Lewis acids like BF3 works in a similar way; coordination to the oxygen lone pair facilitates O-C bond breaking and deprotection. Other Lewis acids like ZnBr2 or MgBr2 can be used for some substrates as well, particularly if two coordination sites are present (e.g., carbohydrates). In these cases, neighbouring group effects with bidentate coordination can be observed.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
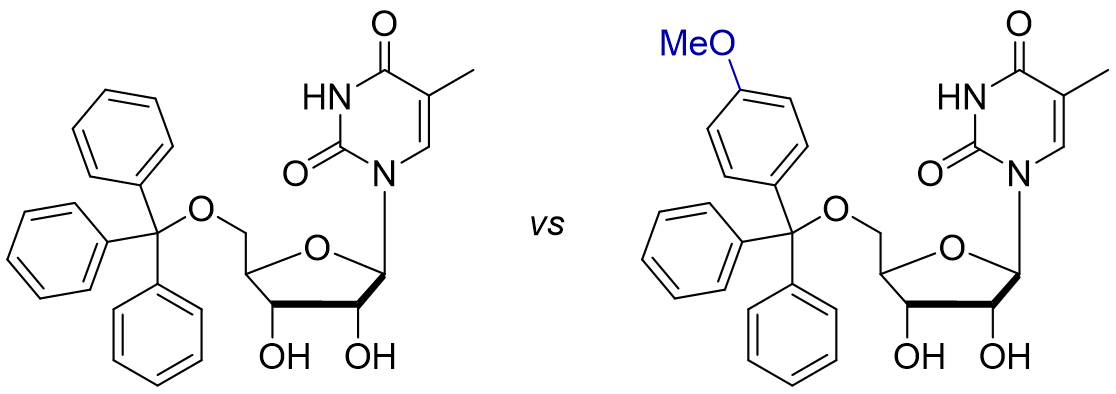
p-Methoxy Trityl Protecting Group
Do you expect the p-methoxy trityl variant to be (more) acid- or base-sensitive?
As a twist on the standard trityl group, chemists have also explored variants such as p-methoxy trityl. A nice synthetic study on nucleotides led to the discovery of such groups – already in 1962 [2]! Their results were as you might expect: By adding a p-methoxy group, we increase stability of the intermediary trityl cation due to the mesomeric electron-donating effect! This makes deprotection easier.
It turned out introducing one methoxy group increased the rate of deprotection by a factor of ten. While standard 5′-trityl-uridine required 48h for complete hydrolysis in 80% acetic acid at room temperature, the mono-methoxy-trityl MMTr group took just 2h! They also developed di- and trimethoxy trityls (i.e., one p-methoxy on each phenyl ring) which cleave in 15min and 1min, respectively. By the way, this change makes initial protection easier too because we also go through the trityl cation.
This di-methoxy DMTr group is one of the most used members of the trityl family due to its reactivity and selectivity for primary alcohols (primarily seen in automated solid-phase synthesis of nucleotides).
A mixture of di-TBS gemcitabine (671 mg, 1.47 mmol) and tritylating reagent (2.94 mmol, 2 equiv.) in dry pyridine (7.3 mL) was stirred overnight at room temperature. Methanol was added to the solution for quenching. After removal of the solvent, the residue was purified by flash column chromatography on silica gel to obtain the title compounds.
Trityl deprotection experimental procedure
Bronsted acid [4]: Compound II (200mg, 0.4mmole) was treated with 3ml of cold formic acid (97+%) for 3 min and then evaporated with an oil pump at room temperature. The residual gum was evaporated twice from dioxane, followed by evaporations from EtOH and Et2O. Finally, the residue was extracted with 10ml of warm H20, the insoluble triphenyl-carbinol was filtered, and the filtrate was evaporated in vacuo. The residual gum was dissolved in EtOH(1ml), dry Et2O (20ml) was added, and the product was precipitated with petroleum ether (30-60°, 10ml) (the gummy precipitate was chilled and scratched to induce crystallization). Recrystallization from the same solvent system gave fine needles of VI.
Lewis acid [5] To a mixture of 4 (2.0 mmol, 994 mg, 1.0 equiv) in CHCl3/MeOH (16 mL/4 mL) was added BF3·OEt2 (4.0 mmol, 0.5 mL, 2.0 equiv) at room temperature. The mixture was stirred at rt for 45 min and was then poured into EtOAc/H2O (100 mL/100 mL). The organic layer was washed with brine (100 mL), dried (Na2SO4), and filtered. After removal of solvent, CH2Cl2 (10 mL) and hexane (30 mL) were added sequentially to the crude product. The resulting solid was filtered and was washed with Et2O/hexane (2/3, 20 mL). The product was dried to give 474 mg of 12 (93%) as a white solid.