Ever struggle with how to identify if a group is a nucleophile vs. electrophile?
If you are in a rush and don’t care about learning chemistry (bruh), the first sections got you. I recommend you try to truly understand the concept as it’s arguably the most important skill in organic chemistry.
- Nucleophiles (nucleus-loving) are neutral or negatively charged species that donate high energy electrons to form new bonds with electrophiles (electron-loving), which are neutral or positively charged species that can easily accept electrons
- Nucleophiles are Lewis bases (e.g. NH3); electrophiles are acids (e.g. H+, BF3)
- The chemical bonding occurs between the highest occupied (HOMO) of the nucleophile and the lowest unoccupied molecular orbital (LUMO) of the electrophile
- Single atoms, sigma-bonds and pi-bonds can be nucleophilic or electrophilic – and the same molecule can contain both (one part nucleophilic, one part electrophilic)
- Nucleophiles and electrophiles are complementary, they react with each other. You will never see a reaction where two groups react with each other as nucleophiles!
Level 0: Chemical bonds are like … financial transactions?
Chemistry is like finance! Simply swap electrons (chemical currency) for money.
If you’re a student, you’re likely broke (you are in need of, and easily accept money => you are electrophilic).
You go to your parents to borrow some money for a dinner (they can donate money => they are nucleophilic).
By getting money and the food, you get happier (you are energetically stable). Your parents are also stoked they can spend some time with their favorite child (they are also energetically stabilized).

This explains how we draw curly arrows, representing electron movement: There are no electrons at the electrophile (it is broke). Instead, the electron donation arrow starts at the nucleophile and points to the acceptor, the electrophile.
Level 1: Nucleophile vs electrophile
You have two options on how to remember which is which:
1. Not recommended: You force-memorize some analogy like above with no brain cell activation, setting you up for nice failures in organic chemistry exams
2. Recommended: You just use your brain and simply remember that “phile” means liking or loving (if you struggle to remember, you know a highly criminal term…)
| Nucleophile | Electrophile | P*d*phile | |
|---|---|---|---|
| What it has | Has more than enough electrons | Has some sort of positive polarization | Problems, chocolate & puppies |
| What it wants | Wants to share its electrons (likes positive charges) | Wants to accept other electrons (likes electrons) | Kids |
| Charge | Neutral or negative | Neutral or positive | Prison sentence |
| Orbitals | High-energy occupied | Low-energy unoccupied | Orbits around kids and playground |
| Acid or base | Lewis base | Lewis acid | Puts them in his base(ment) |
The electrons that a nucleophile donates form the new bond with the electrophile. A simple example is the protonation of water. Which is the nucleophile vs electrophile?

Reviewing the more complex mechanism for the hydration of formaldehyde, you will realize that:
1. Specific atoms or also bonds can have nucleophilic or electrophilic behavior
For example, in the first step, the carbonyl pi-bond is the electrophile. In the second step, the proton H+ is the electrophile.
2. For reactions with multiple mechanistic steps, there can be many different nucleophiles and electrophiles
For example, after the first addition to the carbonyl, the previously nucleophilic water molecule turns into a cationic intermediate which is now electrophilic (which is why it reacts with another nucleophilic water in a deprotonation)

Tired of serious chemistry?
Take a break with “Periodic Tales – The Freshman Mole”, a satirical novel that’s the opposite of educational.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Level 2: Types of nucleophiles
How do we identify electron donors and electron acceptors? There are three categories for both. It’s easier for nucleophiles, so let’s start there.
To donate an electron pair, you need to have an electron pair somewhere (duh).
In our analogy, you need to find the bank vaults that are filled with cash (electrons)!

1A) Lone electron pairs, e.g., water H2O or ammonia NH3 (neutral nucleophile)
Reaction example: Protonation of neutral bases
1B) Negative charges (also electron pairs), e.g., hydroxide OH– or cyanide CN– anions
Reaction example: Basic hydrolysis of an ester
2) Bonding pi-orbitals, most notably C=C double bonds, e.g., alkene or aromatic ring
Reaction example: Bromination of alkenes, electrophilic aromatic substitution
3) Bonding sigma-orbitals with highly electropositive atoms, e.g., methyllithium Li-CH3
Reaction example: Carbonyl reduction with LiAlH4, organometallics
This should make sense. These are the only “ways” you will ever find electrons in a molecule. Either as an unbound electron, in a pi-bond, or in a sigma-bond. This corresponds to the idea of highest occupied molecular orbitals (see below).
Level 2: Types of electrophiles
What about electrophiles? Two categories are similar, and one is different.
To accept an electron pair, you need to have an empty orbital somewhere (duh). Here, we need to find the empty wallets that are eager to get filled with money!
Like nucleophiles, we have pi- and sigma-bonds as acceptors (as they have empty orbitals that can be filled with electrons). However, instead of electron pairs for nucleophiles, we need to look for empty orbitals on single atoms. Think of the empty orbitals like filled purses

1A) Positive charges representing an empty p orbitals, e.g., proton H+
Reaction example: Protonation of any base
1B) Neutral molecule with empty p orbital, e.g., Lewis acids like BF3 or AlCl3
Reaction example: Friedel-Crafts acylation
2) Pi-bond next to electronegative/ stabilized system, e.g., carbonyl
Reaction example: Aldehyde hydration, conjugate addition
3) Sigma-bond to electronegative atom, e.g., methyl iodide CH3-I
Reaction example: SN2 reaction, bromination of alkenes
For both of these categories, I always represented the nucleophile as “Nu-” and electrophile as “E+”. But any combination of these categories works – e.g., an nucleophilic pi-bond can attack an electrophilic pi-bond!
If you know a fair share of reactions, try to think about additional examples for each!
Level 3: Orbitals
Let’s get to the bottom of this (without me writing another text book on orbitals).
Remember that reactions take place if molecules…
1. Overcome their electronic repulsion via charge attraction and/or orbital overlap
(all reactions have activation energies)
2. Have orbitals of appropriate energy to interact
(if orbital energies are too different, there is no energy gain from bonding)
3. Orient in a geometry that allows their orbitals to overlap in a bonding interaction
(if a key can’t get into a lock, the door can’t be unlocked)
Point #2 is critical: energy levels of the nucleophile and electrophile orbitals.
The strongest stabilization comes from interacting orbitals with similar energies.

All molecules have many low-energy, unfilled orbitals and high-energy, empty orbitals (illustrative grey orbitals). We can ignore all because the energy differences between them are too large. The most relevant interaction will be the one between the lowest-energy unoccupied (LUMO) and highest-energy occupied molecular orbital (HOMO).
The higher-energy a HOMO is, the easier it can donate electrons into LUMOs.
The lower-energy a LUMO is, the easier it can accept electrons.
This picture explains the types of electrophiles…
1A + B) Empty orbitals can be LUMOs (neutral or positively charged atom)
2) Pi-bonds always have an empty antibonding pi-star MO which can be a LUMO
3) Similarly, sigma-bonds have an antibonding sigma-star MO which can be LUMO
… and types of nucleophiles:
1A +B) Lone pairs can be HOMOs (neutral or negatively neutral charged atom)
2) Pi-bonds always have a filled bonding pi MO which can be a HOMO
3) Similarly, sigma-bonds have a filled bonding sigma MO which can be a LUMO
Recap: Nucleophile vs electrophile
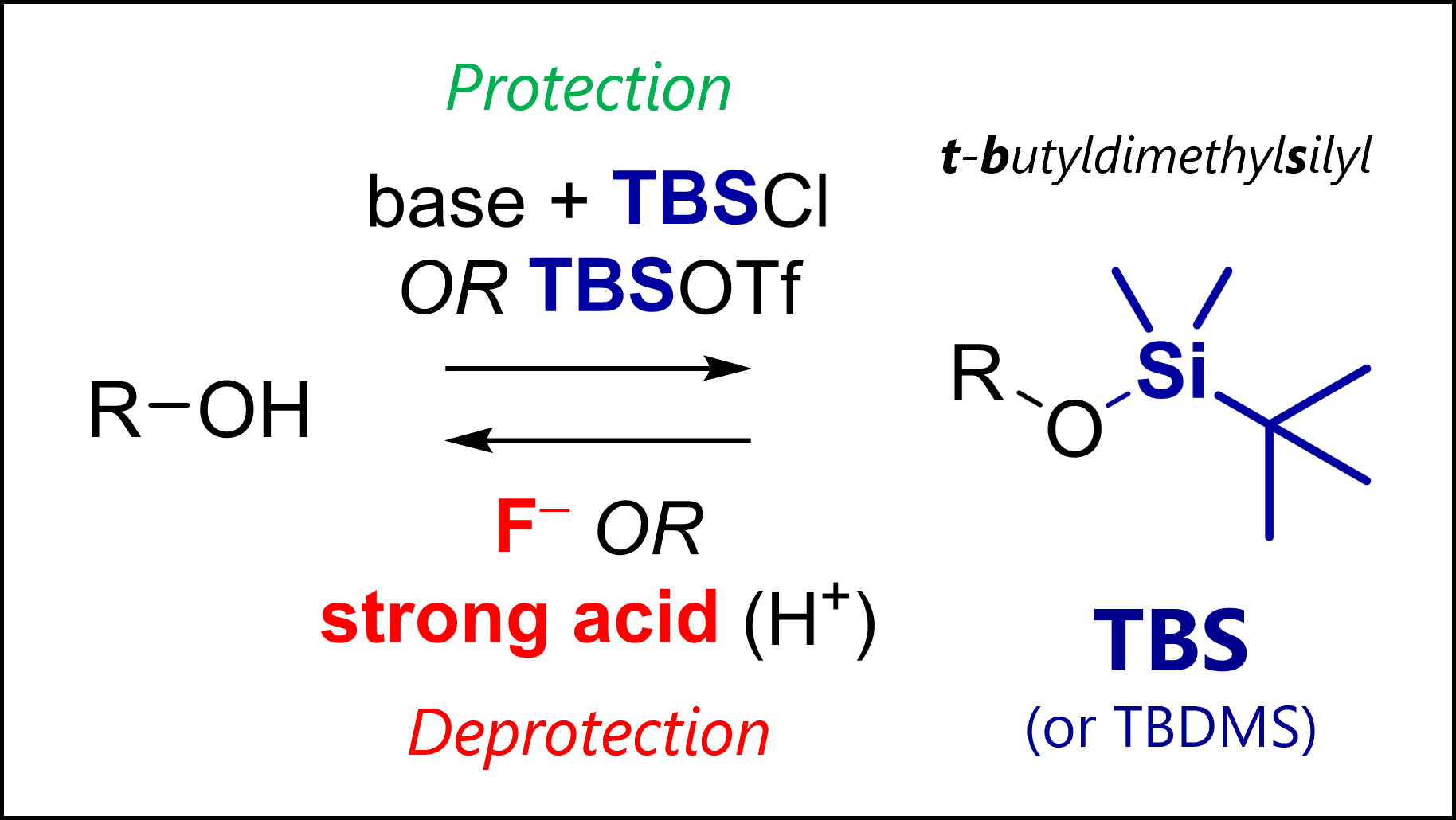
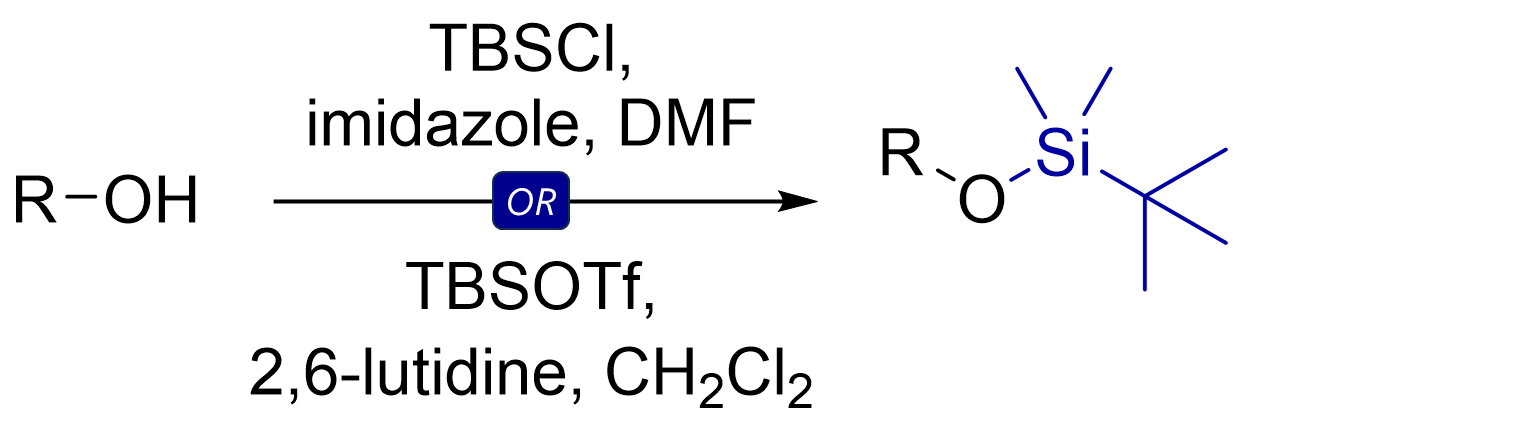
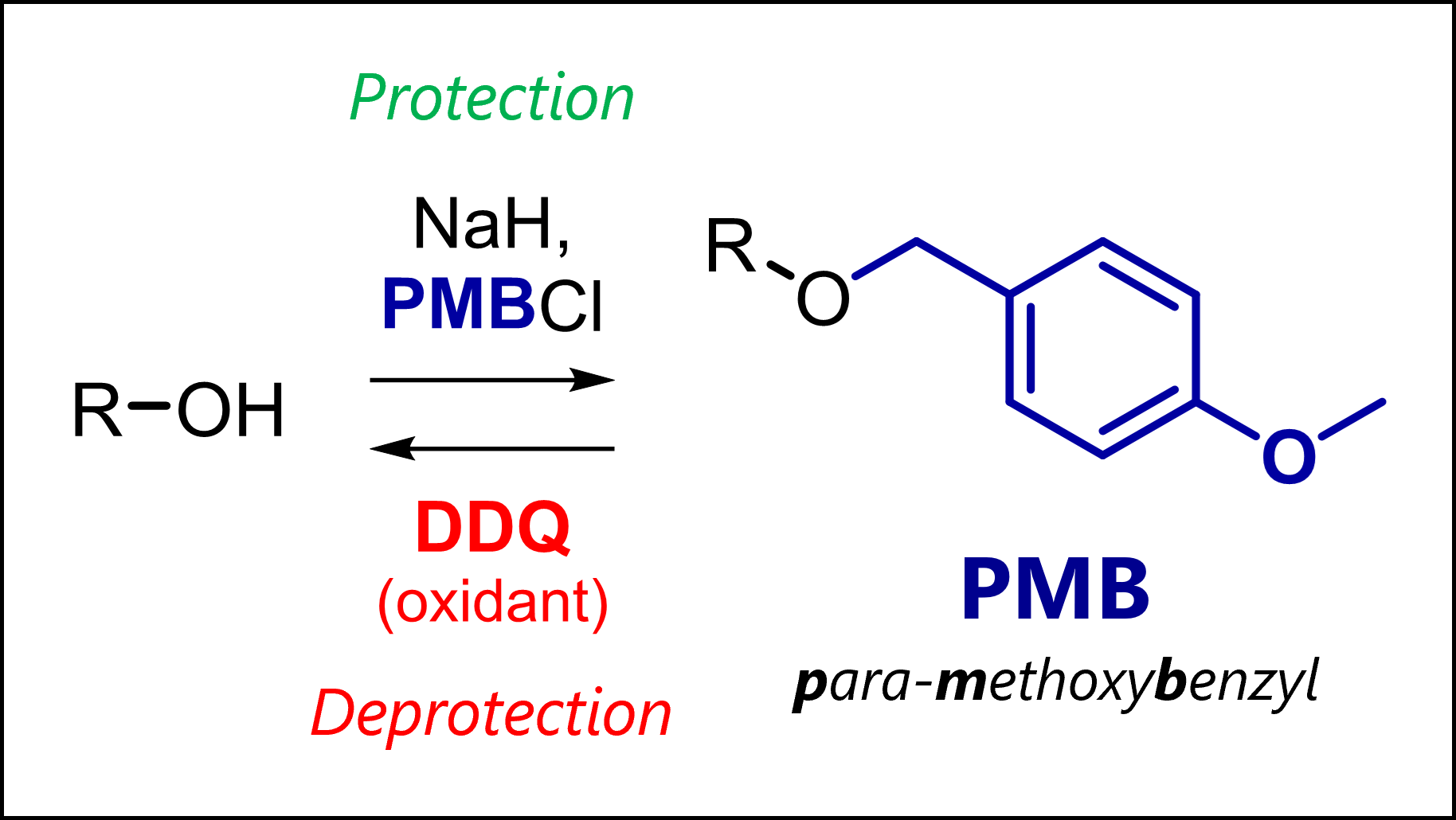
By now you should be able to identify a nucleophile vs electrophile, and know the different types that exist. As an exercise, review reactions you already know or that I discuss on my channel, or functional groups (e.g., protecting groups) to apply your learnings.
There are more nuances and explanations which are not digestible in a single post. So, future posts will explain things like:
– What are the most common nucleophiles and electrophiles?
– How does conjugation to electron-donating or -withdrawing groups influence nucleophilicity or electrophilicity?