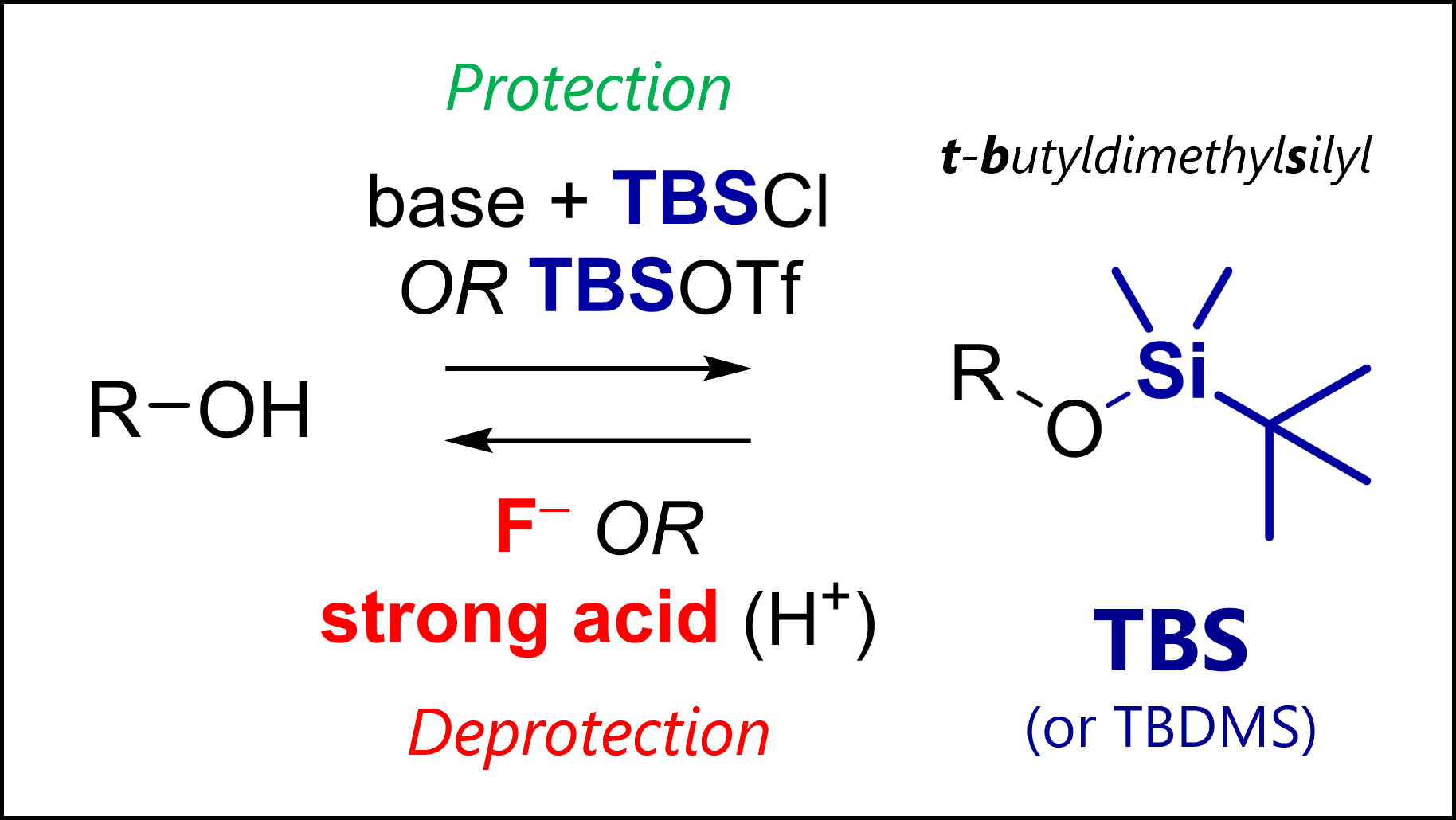
The TBS protecting group protects alcohols in organic synthesis. TBS is deprotected with fluoride anions (e.g., TBAF) or strong acids.
This bulky silyl PG is resilient, allowing for selective/ orthogonal deprotection of similar groups like TMS.
For silyl ethers, bigger is better!
As they have 3 options for deprotection, they’re a nice general learning opportunity for students.
👀 Here’s an interactive 3D model of the TBS protecting group.
What is the TBS Protecting Group?
TBS or TBDMS is short for tert–butyldimethylsilyl, a protecting group for alcohols.
TBS was introduced by the legendary E. J. Corey in 1972 [1] as an evolution to simpler silyl ethers which had already been known. Note this was right around the time of Fmoc discovery, and relatively late in the history of organic synthesis.
The research already covered protection and deprotection conditions still used today. It is one of the most cited JACS publications ever. A decade later, Corey also introduced triflate reagents for silyl ether synthesis [2].
TBS Protection Mechanism
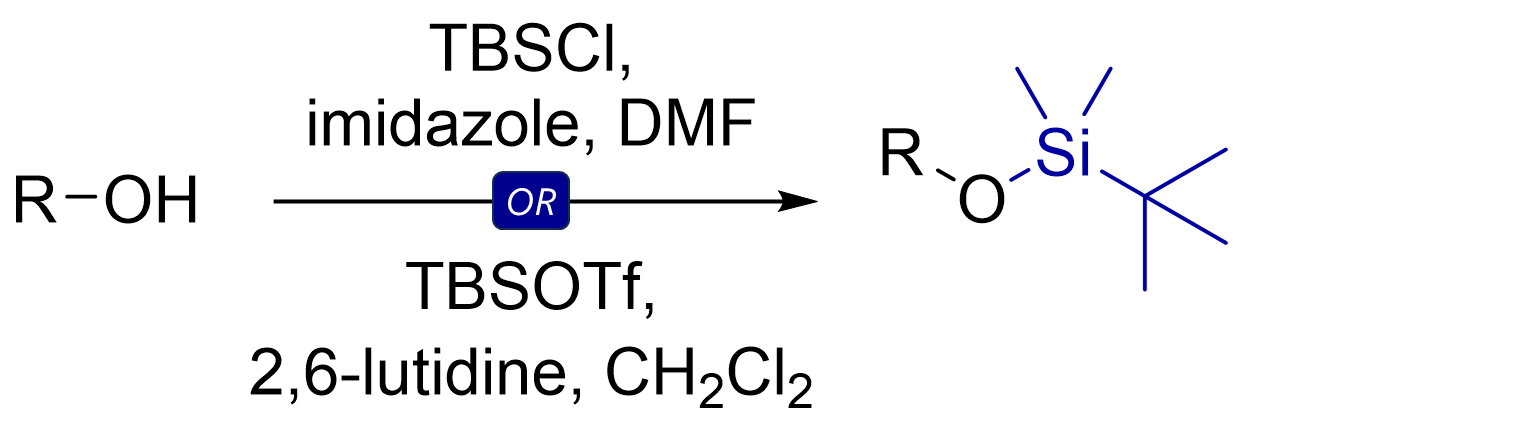
The most common TBS protection conditions are TBS-Cl (forms hygroscopic white crystals) in DMF with imidazole or DMAP.
Corey encountered challenges using TBS-Cl for protection of tertiary or hindered secondary alcohols. Use of TBS-OTf triflate (with 2,6-lutidine as base in solvents like dichloromethane) proved more potent for such cases.

The classic mechanism contains generation of an imidazolium intermediate which acts as a silyl transfer reagent for the actual silylation.
The steps might look counterintuitive. Silicon, in contrast to carbon, can have five bonds at once! Both silylations likely proceed through associative substitutions with pentavalent silicon intermediates. However, you’re likely also fine drawing a concerted step.
Silyl ether protecting group Stability
Many different variants of silyl ethers exist. Despite their overall similarity, they can behave quasi-orthogonal due to different lability. Below, you can see the relative stability towards acidic and basic aqueous hydrolysis.
| Silyl ether protecting group | Stability to acid | Stability to base |
| TMS (trimethylsilyl) | 1 | 1 |
| TES (triethylsilyl) | ~60 | ~10-100 |
| TBS (tertbutyldimethylsilyl) | ~20’000 | ~20’000 (likely higher as basic stability > acidic) |
| TIPS (triisopropylsilyl) | ~700’000 | ~100’000 |
As you can see, bigger silyl ethers are more stable than smaller ones. This stability directly shows – TBDMS ethers are stable to chromatography and survive various reaction conditions which smaller ethers do not. With different labilities, chemists can deprotect TMS or TES ethers in presence of TBS protecting groups (more below).
TBS Deprotection Mechanisms
There are three types of deprotection mechanisms for TBS and silyl ethers in general.

1. Acidic hydrolysis works through protonation of the protected oxygen atom, followed by associate hydrolysis with a pentavalent silicon intermediate.

2. Fluoride-mediated deprotection is based on nucleophilic attack onto silicon, giving a pentacoordinate siliconate intermediate that can release our deprotected product. This is like the imidazole catalysis we’ve seen during TBS protection.
The thermodynamic driving force is the formation of the exceptionally strong Si-F bond (>30 kcal/mol stronger than Si-O) bond. For fluoride-mediated deprotection, TBAF (tetrabutylammonium fluoride) is the archetypical fluoride source. However, other sources like HF or amine-HF complexes are also commonly used.
3. Basic hydrolysis can theoretically also occur with TBS ethers, even though have high stability towards bases. However, at very forcing basic conditions, TBS ethers can hydrolyse (e.g., excess LiOH, dioxane, EtOH, H2O, 90 °C).
The mechanism is also based on nucleophilic addition to silicon, followed by Si-O bond breaking.
Selective Deprotections of silyl ethers
So, we have mentioned that selective deprotection of silyl ethers might be possible. The first category of reactions follows the steric stability we laid out above. Let’s look at some examples [4].

Pyridinium p-toluene sulfonate (PPTS) in protic solvents like MeOH is the mildest system for deprotection of TBS ethers. In this example, you can see that the conditions are controlled enough so that the bulkier TIPS group does not fall off. The same would go for TBDPS (tert-butyldiphenylsilyl).
The next step in the synthesis is addition of K2CO3 to the diol. What is the product?

This next example is sneaky. Don’t be fooled by seeing “TMS” and thinking it will immediately be less stable than TBS! Upon close inspection, we realize that this is actually a alkyl silyl group, part of the so-called SEM protecting group. It can be removed through fluoride anions or more forcing acidic conditions.
Another question: Propose a mechanism the deprotection of SEM with TBAF!
Welcome to chemistry, it’s sometimes random
The second category of selective reactions does not follow any obvious rules. It’s really experimental randomness. A commonly cited example is from the synthesis of zaragozic acid C [5].

During this effort, it was possible to differentiate between two very similar TBS protecting groups. The use of dichloroacetic acid in MeOH was mild enough to selectively (90% yield) cleave only one of the TBS groups.
That same synthesis actually also nicely demonstrated the concept of selective protection based on steric hindrance. After full consumption and protection of the primary alcohol with TBS-Cl, the chemists threw in TMS-Cl to protect the tertiary hydroxyl group as well.

There’s a recent example (2024) of TBS randomness [6]. In the last synthetic steps towards (+)-heilonine, the authors performed a Clemmensen reduction with Zn in HCl to reduce the ketone. It would have been nice if this one-two punch would have directly delivered the natural product. However, for no apparent reason, only one of the two TBS groups fell off. So, a separate deprotection step with TfOH was required.

That’s it for TBS. Learn more about other protecting groups, or watch my educational videos for advanced science content!
TBS Protection experimental procedure [7]
“To a solution of the diol (57.48 g, 0.354 mol) in DMF (354 mL) were added imidazole (96.52 g, 1.418 mol) and TBSCl (160.27 g, 1.063 mol) at rt. After stirring at 50 °C for 17h. H2O was added and the mixture was extracted with Et2O. The organic layer was washed with brine, dried over MgSO4, and evaporated in vacuo. The residue was purified by flash column chromatography (SiO2; n-hexane:EtOAc = 10:1) to give the diTBS ether (138.48 g, 100%).”
TBS deprotection experimental procedure [7]
“To a solution of the alcohol (45.2 g) in THF (350 mL) was added TBAF (1.0 M in THF, 184 mL, 0.184 mol) at rt. After stirring at rt for 18 h, the mixture was concentrated in vacuo. The residue was purified by flash column chromatography (SiO2; n-hexane:EtOAc = 4:6→EtOAc) to give diol (29.1 g, 97% yield, 2 steps).”
TBDMS Protecting Group References
- [1] Corey, E. J.; Venkateswarlu, A. J. Am. Chem. Soc. 1972, 94, 6190 | Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives
- [2] Corey, E. J.; Cho, H.; Rücker, C.; Hua, D. H. Tetrahedron Lett. 1981, 22, 3455 | Studies with trialkylsilyltriflates: new syntheses and applications
- [3] T. W. Greene, P. G. M. Wuts, Protective Groups in Organic Synthesis (Wiley)
- [4] P. Kocienski, Protecting Groups (Thieme Verlag)
- [5] Carreira, E. M., Du Bois, J.; J. Am. Chem. Soc. 1995, 117, 8106 | (+)-Zaragozic Acid C: Synthesis and Related Studies
- [6] J. Am. Chem. Soc. 2004 ASAP | Convergent and Efficient Total Synthesis of (+)-Heilonine Enabled by C–H Functionalizations
- [7] J. Am. Chem. Soc. 2004, 126, 14374 | Total Synthesis of Brevetoxin-B