THCP or THC-P is a recently discovered cannabinoid found in the cannabis plant, joining the ranks of THC and CBD. It has garnered broad attention in cannabinoid research due to its therapeutic potential. There are some wild claims of potency out there. What are the real facts and science behind THCP and how is it chemically synthesized?
What is THCP?
Did you know that almost 150 distinct cannabinoids have been isolated to date? Cannabis sativa is a plant that sparks debates. Some see it as a valuable source of medicine for conditions like glaucoma and epilepsy. But, at the same time, it’s the most commonly used illegal drug worldwide.
At a molecular level, THCP shares the basic framework common to cannabinoids such as tetrahydrocannabinol (THC / delta-9-THC) and cannabidiol (CBD). You can see the chemical structure in the figure.
What sets THCP apart is its heptyl side chain. Chemically speaking, this is a seven-carbon alkyl group with the chemical formula -C7H15. THC on the other hand bears a pentyl group (-C5H11). The length of this chain directly influences binding to the CB receptors(see below) and thus the cannabimimetic activity. This side chain is also called the pharmacophore due to its influence on biochemical activity.
THCP Flower Concentration
The concentration of THC-P in Cannabis Sativa is estimated to be 0.0023% to 0.0136% (0.02–0.14 mg/g) [1]. In comparison, normal THC occurs in up to 30%! These levels seem too low to trigger significant effects or subtherapeutic.
However, not only is THC-P a stronger CB binder than THC, but other phytochemicals may influence efficacy and experience of C. sativa use. Through a so-called entourage effect, there may be synergistic interactions with the major cannabinoids and other phytochemical components.
Due to the low natural concentration, THC-P is more conveniently (for research purposes) synthesized chemically.
How is THC-P Made? (Synthesis)
The organic chemistry behind THCP is actually simple. The starting material is a chiral allylic alcohol for the enantioselective synthesis. The hydroxy group can be substituted with an aromatic ring bearing the linear side chain. Under acidic conditions, we have first the allylic substitution and second an addition of a phenol group to the olefin.
Taken from: Scientific Reports 2019, 9, 20335 (Creative Commons 4.0) Reagents and conditions: (a) 5-heptylbenzene-1,3-diol (1.1 eq.), pTSA (0.1 eq.), CH2Cl2, r.t., 90 min.; (b) 5-heptylbenzene-1,3-diol (1.1 eq.), pTSA (0.1 eq.), DCM, r.t., 48 h; (c) pTSA (0.1 eq.), DCM, r.t., 48 h; (d) ZnCl2 (0.5 eq.), 4 N HCl in dioxane (1 mL per 100 mg of Δ8-THCP), dry DCM, argon, 0 °C to r.t., 2 h. (e) 1.75 M potassium t-amylate in toluene (2.5 eq.), dry toluene, argon, −15 °C, 1 h.
The only issue in this very direct synthesis is the formation of olefin isomers. To get to the right configuration in THC-P, the intermediate (-)-trans-Δ8-THCP can be isomerized. This works through step-wise hydrochlorination and a surprisingly very selective elimination using potassium t-amylate as base.
THC-P Mechanism of Action
THC-P interacts with cannabinoid receptors CB1 and CB2 which are part of the so-called endocannabinoid system or ECS. The ECS is a complex network of receptors, endocannabinoids, and enzymes distributed throughout the body. It plays a crucial role in maintaining homeostasis which is the body’s self-regulation process.
Early research suggests that THC-P may have a higher affinity for CB1 receptors, which are predominantly found in the central nervous system. CB1 receptors play a key role in regulating neurotransmitter release, impacting various physiological functions such as mood, appetite, and pain perception.
Taken from: Scientific Reports 2019, 9, 20335 (Creative Commons 4.0) In vitro activity and docking calculation of Δ9-THCP. (a) Binding affinity (Ki) of the four homologues of Δ9-THC against CB1 and CB2. (b) Dose-response studies of Δ9-THCP against hCB1 and hCB2. (c) Docking pose of (-)-trans-Δ9-THCP (blue sticks), in complex with hCB1. (d) Binding pocket of hCB1 receptor, highlighting the positioning of the heptyl chain within the long hydrophobic channel of the receptor.
The figure d) shows the binding very nicely. The yellow dashed line represents a pocket of hydrophobic amino acids where the linear alkyl side chains reside. Because the longer heptyl side chain has more contacts, it shouldn’t surprise you that THC-P had stronger binding compared to normal THC.
As you can see in a), THC-P has a >30-fold and >6-fold increased binding for the cannabinoid receptor 1 (CB1) and cannabinoid receptor 2 (CB2). The affinity value of 1.2 nM (nanomolar) is very strong on a “molecular” level!
However, the research is too immature to be able to say “how much more potent” it actually is. Nevertheless, preliminary evidence suggests that THC-P may modulate neurotransmitter release in a way that differentiates it from other cannabinoids. There are several areas of potential therapeutic application.
THC-P Effects: Appetite Regulation
One notable aspect of THC-P is its reported appetite-suppressant effects. We clearly need more research to understand the underlying mechanisms but this property raises the possibility of THC-P as a tool for weight management and addressing conditions related to overeating. A study conducted on rodents investigated the effects of THC-P on feeding behavior. The results suggested that THC-P administration led to a reduction in food intake, providing initial support for its appetite-modulating properties.
THC-P Mood Disorders and Anxiety
The interaction of THC-P with CB1 receptors, particularly in the brain, points to potential applications in mood disorders and anxiety management. The modulation of neurotransmitter release in key brain regions may offer a novel approach to addressing conditions characterized by mood imbalances. However, preclinical studies have so far only indicated the anxiolytic potential of cannabinoids in general, not THC-P specifically.
THCP Epilepsy
THC-P may hold promise for neurological conditions. Conditions such as epilepsy and neurodegenerative disorders might benefit from further investigation into THC-P’s effects on neuronal excitability and neuroinflammation. Preliminary data from animal studies has shown cannabinoids, including THC-P, to have anticonvulsant properties.
Is THCP Legal?
The legal status of THC-P varies by country. In the United States, THCP unlike THC is not specifically listed as a Controlled Substance federally. However, regulations vary by state or country. In the rest of the world, THCP is currently legal in Germany, for example. Other countries classify THC-P as a controlled substance. With the current dynamic regarding medical use of cannabis, shifts in legal stands and regulations are to be expected. We have discussed increasing interest on the public level on psychedelic compounds such as ibogaine or psilocybin/ psilocin.
Is THCP Safe?
Given the early days of THCP research, safety aspects, particularly in humans, are not clear. The psychotropic effects of THC-P could raise concerns, particularly regarding cognitive function and the potential for dependence. Long-term studies are essential to assess the safety profile and any adverse effects associated with THC-P use.
In addition, it will be key to establish standardized testing methods to validate high-quality material for research purposes. Standardization is essential for accurate dosing, reproducibility of results, and ensuring the reliability of research.
As THCP is present in only little amounts in C. sativa, THCP products on the market may have been produced synthetically and not have been tested for safety, purity, or potency. Thus, we discourage consumption of (any) supposedly safe drugs and medicines in absence of professional medical oversight and need.
Summary
THC-P, with its distinctive molecular structure and potential therapeutic applications, represents a promising avenue for cannabinoid research. As our understanding of its chemical properties and interactions with the endocannabinoid system deepens, the door opens to innovative approaches in medical science.
While challenges exist, the increasing scientific interest suggest that THC-P could play a significant role in the future of medicine. Responsible research, transparent communication, and thoughtful regulation are paramount in unlocking the full potential of THC-P, other cannabinoids and medicines at large.
Information for THCP
J. Nat. Prod. 2021, 84, 2, 531 | (−)-trans-Δ9‑Tetrahydrocannabiphorol Content of Cannabis sativa Inflorescence from Various Chemotypes
Scientific Reports 2019, 9, 20335 | A novel phytocannabinoid isolated from Cannabis sativa L. with an in vivo cannabimimetic activity higher than Δ9-tetrahydrocannabinol: Δ9-Tetrahydrocannabiphorol
Exercise mimetics: Watch this video or read the written blog below!
Are you tired of dieting or pounding the pavement like David Goggins just to shed a few pounds? Imagine a future medicine that could mimic the benefits of literally running for days.
Weight loss meds like Ozempic have been sending shockwaves through Hollywood and Wall Street. Advocacy by famous figures went viral on social media, causing supply shortages and more recently, questions on their safety emerged. The stonks of select pharma companies exploded, with Danish Novo Nordisk’s market capitalization surpassing the country’s GDP.
Is this first wave of drugs already the be-all, end-all? Side effects like loss of hard-earned gains and pooping pants lead to many users stopping treatment.
Meet SLU-PP-332, a simple small molecule melting fat and effectively mimicking marathon training in mice – all without setting a tiny paw on a treadmill, and without eating less. We will cover this molecule’s discovery, chemical synthesis, and pre-clinical efficacy. This will enrich your interdisciplinary knowledge and give you some practise for data interpretation. We will also explain how other exercise mimetics work and cover random facts, such as taking a closer look at alleged health benefits of red wine.
How obesity Drugs Work
So regardless how we feel about it, obesity is the problem for healthcare systems, and source of many problems. By 2030, nearly half of Americans will be obese – not to mention overweight.
Why even consider drugs for weight loss? We all know that exercise and diet regimens have very low compliance in reality. People just don’t stick to them even if they know they should. Also, some patients have co-morbidities that make them exercise-intolerant. It doesn’t matter how much you want to be David Goggins – if you have chronic heart failure, you can’t run for days.
Another issue: the emerging obesity drugs melt away body mass, but much of it is also muscle tissue. Drugs which can trigger fat loss, retain muscle and simulate exercise would be helpful for many people.
Obviously, with hundreds of millions of obese people, this is a massive long-term opportunity for pharmaceutical companies. This bullish outlook has resulted in strong investor interest in obesity drug developers, adding billions of dollars to their valuations.
The drugs behind this gold rush are GLP1 agonists. As mimetics of the incretin hormone GLP1, they stimulate insulin production and help manage blood sugar – this is especially key for diabetics. They also slow down movement of food in your stomach which can help patients feel fuller faster and curb hunger. Leave a comment if you want to learn more about these hyped obesity drugs in a future video – they have a massive history of research behind them. Today, we will instead check out the so-called exercise mimetics.
Science Behind Exercise Mimetics
PGC-1a is a key link between endurance exercise and physiological adaptation. Expressed in various tissues, this is the master regulator of creation of mitochondria – also known as the powerhouse of the cell – as well as other processes like glucose and lipid metabolism. Because it’s a coactivator, it interacts with other transcription factors to modulate the expression of certain genes. Looking at the example of detoxification of reactive oxygen species, we realize this gets into complex cellular biology territory. Due to this complexity, dysregulation of PGC-1 alpha disrupts physiological processes and contributes to many diseases.
Why is this relevant for exercise mimetics? Well, while various mimetics have different primary targets, most ultimately all trace back to PGC-1 alpha.
One rather famous molecule in this class is resveratrol. This polyphenol is present in many foods and wines, and it can trigger just about every effect under the sun. It likely indirectly activates the so-called SIRT1 protein, which in turn deacetylates PGC-1 alpha and ramps up beneficial activities. There’s a lot of literature on this if you want to check out the cellular biology.
Some of the first insights came from a 2006 study looking at daily intake of resveratrol in mice being fed a high-fat diet. If you are good at playing ‘spot the difference’, you will notice that fat and muscle tissues feature much denser mitochondria. It looks like these mice adapted to exercise they didn’t perform – thus, we’re calling such effects mimicry.
Shockingly, some human studies showed resveratrol actually blunted some aspects of training adaptation. Ironically, removal of reactive oxygen species by resveratrol might limit training-induced adaptations. This once more highlights that in biology, nothing is simple or black-or-white.
Based on the immature human data, the verdict on resveratrol is still open. If you check Wikipedia, you can see that no health benefits have clear evidence. Such lacking clinical data is a common theme for exercise mimetics in general, as they represent a new class of compounds.
Even big pharma companies dabbled in this space, with GSK paying 700 million dollars for a biotech working on a resveratrol formulation 15 years ago. They did not test it in obesity but rather haematological cancers. This proved to be bad luck as they killed the program after seeing increased risks of kidney failure. You can see that even introducing seemingly healthy substances like resveratrol into medical practice can be challenging.
Estrogen-related REceptors and SLU-PP-332
So let’s get into exercise mimetics more deeply. To understand SLU-PP-332, we need to take a look at another investigational compound. Unlike resveratrol which targeted SIRT1, this one activates the estrogen-related receptor gamma.
This is one of three siblings of the ERR family, expressed in tissues with high energy demands. These nuclear receptors have received considerable attention for their potential value in treating metabolic diseases. As a side note, nuclear receptors are proteins chilling in the cytosol or nucleus. They can sense specific ligand molecules and in turn, regulate expression of specific genes.
As their name suggests, ERRs are structurally related to estrogen receptors (ERs). These nuclear receptors utilize estrogens as ligands and contribute to breast and other cancer types. A key ER-drug is Nolvadex – more famously used by bodybuilders to manage their gynecomastia. Back to ERRs, which despite their resemblance work via different mechanisms than ERs.
The company GSK developed the first small molecule ERR agonists already in the early 2000s. Remember for later that this hydrazone-based agent strongly activates beta and gamma, but not the alpha ERR isoform. ERR beta is a negligible player given it’s not present in skeletal muscle .
ERR gamma highly expressed in oxidative slow-twitch muscle tissues in the calves, with minimal expression in quadriceps which appear more white. Its powerful effects can be clearly seen if a spooky experiment is performed, creating transgenic mice that express ERR gamma more broadly. These super mice have deep red muscle bellies due to improved oxidative capacity, increased vascularization and bigger mitochondria. In an endurance exhaustion test, transgenic mice ran roughly 1500 meters instead of measly 600m by wildtype mice. This means without any specific training, ERR overexpression creates endurance monsters that can run more than twice as far.
We also need to look at ERR alpha, the receptor which was not significantly activated by the GSK compound. Like the related gamma isoform, it’s expressed in skeletal muscle and has similar functions.
We’ve just seen how transgenic mice expressing ERR gamma are endurance monsters. For ERR alpha, scientists also looked at the opposite model – so called knockout mice lacking this important nuclear receptor. These mice are able to live somewhat normally which means that this receptor type is not vital for life. However, if you look at the relative size of the heart and muscles compared to body weight, the knockout mice in blue have significantly lower muscle mass.
As you might expect, this means the mice have lower endurance capacity and reach exhaustion much faster. The realization here is that if lacking ERR alpha results in endurance weakness, we could be able to mimic endurance exercise by activating it with a drug.
SLU-PP-332
The first question is, how do we find an ERR alpha drug? One way is to start with the GSK compound – but wait, didn’t we say this one only activated the beta and gamma ERRs? I’ll explain. First, you have to know that the only available X-ray structure for this molecules is with ERR gamma. This tells us with high certainty what the binding mode looks like – for example, in red we can see that the phenolic hydrogen is involved in a hydrogen bond with an aspartate residue of ERR gamma. This structure can guide the simulation of how the slightly different binding pocket of ERR alpha would bind to ‘4716. As we’ve said the binding is not that strong, but we can use it as a starting point for the design of more potent drugs.
The scientists behind this research identified a crucial phenylalanine at position 328, here in pink, which is present in ERR alpha but not gamma. By engineering interactions with this unique group, we could design a drug that selectively targets alpha over gamma.
This was achieved very easily by converting the iso-propyl benzene of ‘4716 into a naphthalene ring. As you can see from the new simulation, this extended aromatic system can undergo pi-pi stacking with then phenylalanine. This simple change increases affinity for ERR alpha by more than 50-fold. Let’s compare it again to ERR gamma. As the phenylalanine is not present, the interactions are weaker here and the agent is around 4-to-1 selective for the alpha receptor.
The chemistry behind this is so easy that it can be managed by even the clumsiest undergrad . The starting materials are simple and cheap – the only thing needed is cooking them up in toluene overnight. The highly nucleophilic hydrazide adds to the electrophilic aldehyde, creating an adduct. After a proton shift, the intermediate can eliminate water, forming the hydrazone linkage of the product. As it precipitates from the solution, it can be easily separated and subsequently recrystallized to give pure SLU-PP-332.
Dedicated to every chemistry and STEM student who asked: “Why did no one warn me?”
Exercise mimetic effects
By now you are eager to hear about its effects – is it really as impressive as the clickbaity title? Let’s start from micro and go to macro. For some of these, feel free to pause and take more time to digest the info.
Upon treatment of isolated myocytes or muscle cells, the researchers observed a doubling of the maximal mitochondrial respiration rate. Obviously, more oxygen means a higher energy production. Not only are the mitochondria more productive, but there are more of them!
There were also structural differences. Here you can see stained sections from quadriceps muscle. Notice the difference? There’s significantly more green color which corresponds to myosin protein in type IIa fibers which are fast, aerobic muscles. On the other hand, there are lessred, type IIb fibers. These are muscles which act fast but use anaerobic metabolism, meaning they fatigue quickly. Interestingly, no difference was observed for slow aerobic type I muscle.
Knowing this, what do you expect regarding exercise performance? Well, mice treated for a few days with this compound showed superior endurance without any training, being able to run roughly 70% further than normal mice. Unfortunately, the experimental procedure for this assessment is less fun. You can tell because the wording says “mice were run”. If they subsequently didn’t react to electrical shocks, you know that they were legitimately exhausted.
There are additional investigations into the “why” behind this such as specific gene expression targets. I leave this topic for interested nerds to check out on their own.
One interesting finding was that extended dosing for 2 weeks led to difference in grip strength as well. Unfortunately, the authors don’t describe this in more detail. It looks like grip strength decreased over time for both the active and the control group. This might be because of accumulating fatigue or other things. It would be cool if the authors are correct – meaning if SLU-PP-332 could enhance some strength endurance, and not just pure aerobic performance.
We haven’t covered one major, obvious question yet. The molecule’s exercise mimetic effects are very intriguing, but does it have any impact on weight?
Well, in another recent study the team documented that mice treated with the drug usedmore energy while consuming the same amount of food! Numerically it looks like they even had less food.
Metabolically, ‘332 triggered a shift towards fat burning. Fatty acid oxidation increased by roughly 25%, while the use of carbs decreased reciprocally. Again, more details can be found in the paper.
The magnitude of weight loss depends on the starting point. Normal mice with healthy diet and weight did not lose weight. On the other hand, obese mice eating an unhealthy high-fat diet saw 12% weight loss after one month. The researchers also looked at a genetic model. These mice don’t produce the key metabolic hormone leptin, leading to excessive hunger and food intake. Similar to the diet-induced obese mice, these chunky fellas also dropped significant weight.
We’ve noted that providing more than just weight loss is the next frontier. SLU-PP-332 might be one step in that direction. More pre-clinical work is required to understand its long-term effects. Maybe, related, optimized molecules could be even more potent. Researchers have not seen any safety signals but tolerability, administration and translation will be key to elucidate prior to first-in-human trials. These drugs will (likely) not be launched earlier than the end of the 2030s but with the excitement around obesity, it’s definitely going to remain an interesting space.
References on exercise mimetics
PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism | Oxidative Medicine and Cellular Longevity, 2020, 1452696
Caloric restriction and exercise “mimetics’’: Ready for prime time? | Pharmacological Research 2016, 103, 158
Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha | Cell 2006, 127, 1109
Distribution and Effects of Estrogen Receptors in Prostate Cancer: Associated Molecular Mechanisms | Frontiers in Endocrinology 2022, 12, 811578
Identification and structure-activity relationship of phenolic acyl hydrazones as selective agonists for the estrogen-related orphan nuclear receptors ERRbeta and ERRgamma | J Med Chem 2005, 48, 3107
Exercise and PGC-1 alpha-Independent Synchronization of Type I Muscle Metabolism and Vasculature by ERR gamma | Cell Metabolism 2011, 13, 283
Estrogen-related receptor-α coordinates transcriptional programs essential for exercise tolerance and muscle fitness | Mol Endocrinol 2014, 28, 2060
Watch the video on YouTube or read the written post below!
When people with an IQ of 50 hate on the “evil” scientists of big pharma, they often overlook that there is relentless work and ingenious brainpower behind the discovery and optimization of medicines, including antiviral drugs. If drug discovery would be so easy, pharma companies would not spend up to billions of dollars to get just one drug to the market. Also, chemists would be balling instead of complaining about bleak career opportunities on Reddit.
In this post, we will look at the educational drug discovery journey of an antiviral drug. Just by looking at today’s molecule, you should know this is going to be a nice one – and yes, that’s a boron atom in a pharmaceutical. You will learn why using boron in drugs can be powerful, and why it’s not good if your people in clinical trials turn yellow.
Hepatitis C: Significant Innovation on Major disease burden
Hepatitis C is a severe infectious disease, leading to liver disease and serious complications. The hep C virus chronically infects over 170 million people or over 2% of the world’s population! The dilemma is that disease incidence and drug market are inversely related. The key Western Pacific, Southeast-Asian and African regions have the highest prevalence with 130 million infections and no access to HCV drugs. The US and Europe on the other hand have around 15 million infections but are the target market for drugs. HCV occurs in 6 different “genotypes” or variants across the globe which complicates treatment. High-income countries have primarily the genotype 1, which is only 10% of disease burden in low income countries. Without being a scientist, you can guess that these viral genotypes influence drug sensitivity – that’s also what we’ve seen with C19 vaccines and variants such as Omicron. Oh yeah, and there’s a problem of viral resistance.
HCV spreads via blood-to-blood contact, so injection drug use, poorly sterilized medical equipment and other pathways. In contrast to HIV or Hepatitis B, it is not a STD. An infected individual can show no symptoms for decades while increasing their risk of liver failure and cancer. The good news it that unlike infections with HIV or Hepatitis B, HCV is curable. However, while there are approved vaccines for Hep B, only few are still in development for Hep C. Early physiological studies earned the Nobel Prize in 2020, and the last two decades of saw a true revolution of HCV drug discovery.
As HCV is a viral disease, there are multiple potential inhibition points in the virus lifecycle – entry into the host cell, protein synthesis steps and packaging and release. However, if we contrast HCV drug development with HIV, it becomes evident that there was quite the slow start. While the first antiviral drug was approved 3 years after the virus was identified, it took 24 years from HCV discovery to approval. There are many reasons for this – low perceived market value, low pressure from patient associations but also a poor understanding of the nature of HCV as a disease. Without going through all of the details, advancements in structural biology, pharmacology and also clinical trial design ultimately led to the approval of various therapies.
If you squint hard enough, you might be able to read something!
Most are combinations based on interferon proteins – which work by increasing immune defense – administered by subcutaneous injection, and ribavirin – which is an oral broad-spectrum antiviral that is used against various viral fevers. This “one size fits all” had a decent efficacy of around 55% – but true improvement came only with the development of combination stacks which added a direct-acting antiviral drug. These were more targeted and robust against resistance and also came with a halved treatment duration. As the third evolution, all-oral combinations were also developed – such as the last entry.
You might think, we have more than one blockbuster antiviral drug already on the market that cured millions of patients, why should we look further into HCV? Well, all the remaining, untreated, tens of millions of HCV infected individuals can’t afford outrageous prices of up to over $50K per treatment. Neither can the WHO, who aspires to treat 80% of diagnosed populations by 2030. The development of new direct-acting antivirals thus may serve to increase market competition and lower costs, thereby enabling more equitable access. Also, many of the drugs developed were not equally effective against the six genotypes we covered at the start – so there is more work to be done.
Design of aN antiviral drug against Hepatitis C
We already mentioned that various steps in the viral lifecycle can be potential drug targets. If the virus can’t productively infect host cells, the infection will not be sustained. In 2014, GSK published studies investigating inhibitors of theNS5b polymerase, the viral RNA printer. This is the same mechanism of action of Gilead’s Sovaldi.
Inhibitors can directly block active sites of enzymes – so where the catalysis is occurring – or instead, bind to other, allosteric sites, inducing macro-conformational changes that decrease an enzymes activity. In the case of NS5b, there are four well established allosteric sites – and the palm II site is particularly interesting because it is closest to the active site, and there are many amino acids that are highly conserved HCV genotypes – so a potential inhibitor could combat all variants equally effective.
The GSK team found an inhibitor here tagged with the number (3), which nicely docked into this palm site. This impacts the enzyme in two ways: firstly, the allosteric binding of compound 3 stabilizes a so-called closed state which is much less active. Secondly, the head group of the inhibitor also interacts with catalytic Mg2+ cofactors and thereby disrupts placement of incoming nucleotides, making polymerase initiation and propagation more challenging.
So what do these inhibitors look like? Their quest started from this scaffold – a benzofuran core with a cyclopropyl group swagging around – which was published by researchers from another company. Looking at a terminal alcohol as the “head group” at the sulfonamide, they observed strong activity against wildtype genotype 1 HCV, as well as against a common mutation which occurs at the 316 aminoacid position. The activity was assessed by looking at cell-based replication systems, as well as more “raw” data from a biochemical polymerase assay.
Boron in PharmaceuticaLS?
As mentioned previously, the apolar part of the inhibitor is bound in the grey palm site while the head group looks into the polar active site. By screening through different head groups, the GSK team found that by appending a boronic acid pushed activity into the single-digit nanomolar range, even for the critical polymorph 316N. For you morons out there, the lower the concentration, the stronger the inhibition. Aryl boronic acids were also more effective, particularly for wild type 1a. In contrast to the para isomer, the meta substituted aryl boronic acid showed reduced activity – highlighting the need for proper orientation of the head group.
What about the need for boron? Well, while substituting the boronic acid with other polar groups maintained strong IC50 in the assay in the right-column, it led to significantly lower replicon activity. This is because anionic compounds suffer from lower cell-permeability – due to the low acidity of boronic acids, this problem does not occur for this series. However, when looking at drug metabolism and pharmacokinetics, DMPK, they found that these aryl boronic acids had very low bio-availability in their rat model, and were rapidly cleared and excreted. This challenge was significantly improved by adding a fluorine substituent to the phenyl ring – increasing bio-availability 5-fold, while even further increasing activity, particularly against the 316N variant. They tried to further optimize activity by embedding the boronic acid into a ring – while this resulted in better performance in the polymerase assay, especially the 316N efficacy decreased.
Although less active, this analogue has an easy but instructive synthesis that nicely tests your understanding of fundamental reactions. First, this starting material was carbonylated – of course, this only touches the aryl bromide as fluorides usually don’t react with Palladium. Next, a Sandmeyer reaction exchanged the amine into a new aryl bromide, going through a diazonium as an intermediate. Then, radical bromination installed the benzylic bromide which was coupled with the free sulfonamide of the apolar core. An easy borylation again leveraged the aryl bromide as a functional handle. The final cyclization is triggered by reduction of the ester with Lithium borohydride – which is quite cool.
By the way, if you are wondering about the use of boron in pharmaceuticals. The application of boron in medicine dates back to the early 19th century, when boric acid, so B(OH)3, was used as a mild antiseptic. However, boron derivatives were long neglected thereafter as a result of largely unfounded claims that they are unstable and toxic. After the approval for bortezomib in 2003, there has been quite an upsurge in interest and we will further explore why it can be quite powerful.
Back to the medicinal chemistry: After feeling satisfied with the activity, the team performed crystallizations to reveal binding modes. There are various supramolecular interactions at work – quite basic apolar and polar interactions, but also cation-pi interactions of the electron-rich benzofuran with the positively charged Arginine side chain in the protein. This Arginine is conserved across all HCV genotypes and is the reason why this scaffold is well-suited to occupy this position. This even has a ripple effect as this Arginine anchors a network of hydrogen bon interactions to other parts of the inhibitor. Notably, the boronic acid did not form any covalent bonds or complexes – instead, it forms a hydrogend bonds with a bound water molecule, as well as other polar interactions that are not indicated here.
Optimization and Structure-activity relationship Of the Antiviral Drug
So hey, we got ourselves the final antiviral drug at hand already – right? Well, the molecule we flashed at the very start actually looks slightly different from the last lead compound we saw. Why aren’t we done yet? You see, when the team moved forward to human studies, they saw that the antiviral drug had a very short half-life in blood plasma of 5 hours, resulting in a higher anticipated daily dose for efficacy. Additionally – back to the importance of DMPK – they found that metabolic breakdown resulted in a major metabolite with long half life. This compound was also observed in Phase 2 of another drug, and associated with strong liver toxicity. Although there was no direct evidence of toxicity, the team wanted to avoid its formation to limit adverse reactions and improve its pharmacokinetic profile. The key step to prevent was oxidation of the benzylic carbon – quite evident given that the team could also detect the carboxylic acid product.
To maintain a similar binding mode and not risk starting from scratch, they hypothesized three major approaches that might reduce the propensity for benzylic oxidation. The first two consisted of cyclization, either onto the phenyl ring or in the direction of the sulfonamide moiety, while the third simply excised the benzylic CH2 group. This shortening of course came with the risk of severely disrupting the binding of the head group due to its positional shift. You might think – what the heck, why are we doing these complicated things – can’t we just throw a methyl group on the benzylic position and hope that steric shielding reduces the rate of oxidation? You would be correct – but unfortunately, the authors found that adding a methyl group reduced potency 50-fold. This was probably because the Methyl substituent induces a unfavorable conformational twist – and that’s the authors envisioned the two cyclization approaches to strive towards a more pre-organized conformation.
Let’s look at their results. As you can see in this table, cyclization onto the aryl ring actually reduced inhibition significantly, particularly for 316 variants. So, no good.
Within approach B, the team replaced the sulfonamide with carbonyl containing groups because it appeared that only of the oxygens made meaningful contact with the NS5b protein. The results were mixed – for some analogs, even wild-type inhibition decreased significantly. However, the oxazolidinone 25 showed very good potency. Just again demonstrating that the boronic acid is critical, the team found that removing it resulted in an over >100-fold loss of activity.
So, compound 25 was quite encouraging – and the team figured shifting the ring to an aromatic system should be even more metabolically stable, improving the clinical profile of the molecule. They found that the triazoles, such as compound 31, were basically as potent as the previous oxazolidinone. Removing one of the nitrogens decreased activity significantly as it removed a critical hydrogen bond with the protein. Notably, adding substitution to the triazole diminished activity, highlighting the steric constraints in the pocket. In summary, these aromatic designs were not really better – so the team also looked at the approach C – directly eliminating the culprit, the benzylic CH2 group.
Binding Mode of the antiviral drug
This series was right on the money. Recognizing that shortening of the head group would change the position, the team also looked at the meta-boronic acid – but the para-substitution continued to perform better. Here you can also see the dramatic impact of electron-withdrawing groups on activity, particularly on 316N and 316Y genotypes. But the chloro compound 47 was even more potent – this is why they moved away from the fluorine. The final refinement was closure to a benzoxaborole to enhance chemical and metabolic stability – and thankfully, this modification did not lower the activity too much.
Looking at crystal structures revealed an extensive network of interactions, mediated by highly ordered water molecules – the 3 red balls – which the authors did not observe with other series. Here we see the beauty and complexity of supramolecular interactions – the oxaborole moiety interacts directly with two water molecules: One contacts the backbone N–H of a glycine and the second H-bonds to another ordered water molecule bridging an arginine and asparagine. A close look at the boron reveals that its trigonal planar geometry is slightly distorted – it looks like the proximal water is well-positioned to occupy the empty p-orbital of boron and induce a more tetrahedral configuration. The distance between boron and water is 2.5 Angstrom, which is close enough for a strong interaction but not as short as predicted for a covalent bond. This setup is basically an equilibrium between a water-bound and unbound boronate complex. This interconversion of planar to trigonal binding, leading to multiple potential binding modes, is why boron is such a powerful and flexible functionality.
The final question was to check whether this chemical optimization was actually reflected in an improved pharmacokinetic profile of the antiviral drug.Looking at PK in rat, they found that compounds 33 and 49 had much lower in vivo clearance, and were much more bio-available. Looking into drug metabolism, the team also compared cytochrome p450 inhibition. These enzymes are the major route of elimination for multiple drugs and their disruption is one of the most common mechanisms leading to harmful drug-drug interactions and side effects. For example, analog 33 had a micro-molar activity versus major CYPs, raising a potential risk for clinical development. On the other hand, lead compound 49 was less active, posing significantly less risk.
This improved profile was demonstrated when in first-in-human studies, where they saw much longer drug half life and no oxidation to the potentially toxic metabolite which triggered them to re-explore their strategy. GSK then progressed this asset to phase 2, combining it with an RNA-based treatment called RG-101 of Regulus Therapeutics. After they saw two cases of seriousjaundice – so patients turning yellow – the FDA put a hold on RG-101 and GSK actually also decided to not further develop this compound. Maybe not the success story we were all expecting.
I hope you learned a thing or two from this story. See you next time!
References on Hepatitis C Antiviral Drug Discovery
Design of N-Benzoxaborole Benzofuran GSK8175—Optimization of Human Pharmacokinetics Inspired by Metabolites of a Failed Clinical HCV Inhibitor: J. Med. Chem. 2019, 62, 7, 3254
Discovery of a Potent Boronic Acid Derived Inhibitor of the HCV RNA-Dependent RNA Polymerase: J. Med. Chem. 2014, 57, 5, 1902
HCV796: A Selective Nonstructural Protein 5B Polymerase Inhibitor with Potent Anti-Hepatitis C Virus Activity In Vitro, in Mice with Chimeric Human Livers,and in Humans Infected with Hepatitis C Virus: Hepatology 2009, 49, 3, 745
Ibogaine has built a reputation as an anti-addiction magic bullet. Even the Wolf of Wallstreet is vouching for it, lol. While drug manufacturers are settling lawsuits, the state of Kentucky recently announced they might use double-digit millions for ibogaine research.
Maybe you’ve heard of syntheses and promising effects of psychedelics psilocybin, LSD, THCP or MDMA. This will be just as interesting!
However, the clinical development of psychedelics is not as rosy as some of you might expect. There is an increasing number of case reports with severe and even deadly adverse events at high doses. Thus, scientists pursue next-generation molecules that unify life-changing efficacy with superior safety.
Join me on a journey to learn the biochemistry, therapeutic promise, and chemical synthesis of ibogaine and psychedelics-inspired medicines. How can we even know if these drugs might help, let’s say, heroin addiction?
Let’s start with the basics. What is ibogaine? Iboga comes from the bitter root bark of the Tabernanthe Iboga rainforest shrub native to West-Central Africa. Beyond traditional medicine, iboga also has a long-rooted – pun intended – importance to spiritual practices. From a Western perspective, its ritual use was first documented by French and Belgian explorers in the 19th century. Early on, high iboga doses were shown to induce powerful states of mind but also have toxic side effects. On the other hand, tribal hunters used much smaller quantities as mild stimulants. These guys were already microdosing before it was cool.
History of ibogaine and context:
Its recent history is reminiscent of other substances as it also meets what you could call the “20th century psychedelics starter pack”. Was iboga once sold commercially as a dubious extract, just like psilocybin or heroin? Check. Did the CIA run unsettling experiments as we’ve seen with LSD, in search of agents for warfare or mind control? You bet. And did the FDA classify ibogaine as a devilish Schedule 1 drug – to the dismay of people like Howard Lotsof who started to report anecdotal evidence of potent anti-addictive effects? Check. Although it was indeed abused by athletes as a doping agent, this classification dealt a blow to ibogaine investigations. While some early clinical studies were funded in the 1990s, many were terminated, and progress was quite sluggish.
Before we can understand medicinal effects, we need to take step back again from history. Iboga bark is not a pill, so it contains numerous natural products. This table from a mass spec study just shows ones over 1% – so the full list is long. Like we’ve mentioned for psilocybin, it could be that some of these phytochemicals support some sort of entourage effect of iboga. As the major alkaloid present with 2% of total bark weight, ibogaine is our primary molecule of interest. Here’s a fun fact some of you might find interesting: iboga even contains yohimbine, an alkaloid used as a dietary fat burning supplement.
Biochemical Effects of Ibogaine
In the body, ibogaine has a half life of roughly 7 hours. After ingestion, metabolization through a demethylation kicks in, catalysed by several cytochrome P450 enzymes. The resulting noribogaine with the free phenol group is more persistent. With an even longer half-life, it’s quite evident why ibogaine usually results in psychoactive effects over 24 hours, longer than most other psychedelics. Despite intensive research, we still do not understand these molecule’s mechanisms properly.
I mean, just look at this table – I’m you will agree that it seems complex! Unlike psilocybin or LSD, ibogaine does not get its hallucinogenic properties due to serotonin 2A receptor activation. This sets ibogaine apart from classical psychedelics.
Noribogaine displays sub-micromolar agonistic affinity to the kappa opiod receptor. This profile is reminiscent of the hallucinogenic natural product Salvinorin A, present in the leaves of the Mexican Salvia plant.
Noribogaine is also a strong partial agonist of the related mu opiod receptor – this is the target of classic opiod analgesics such as morphine and fentanyl, commonly used as sedatives or to treat severe pain. These agents are usually very dangerous, highly addictive substances – they are behind the extensive opioid overuse in the US. But as we will see, due to the breadth of molecular mechanisms implicated, ibogaine-derived substances could be helpful in overcoming opioid dependency.
Another key mechanism is the inhibition of NMDA-receptors, similar to drugs like ketamine and even alcohol. This might explain the dissociative effects of ibogaine shared with these other agents. NMDA receptors are glutamate-gated ion channels which drive neural processes like learning, memory, and neuroplasticity. I’m not saying that randomly taking drugs can help neurodegenerative diseases like Alzheimer’s. It’s probably counterproductive, but there is a molecular link here.
I wanted to highlight two other mechanisms – firstly, inhibition of serotonin and dopamine transporters. The 2 micro-molar Ki value for ibogaine and Noribogaine essentially match the affinity of amphetamines. Ibogaine differs from these notorious drugs of abuse as the serotonin uptake inhibition is non-competitive. This and other reasons are why ibogaine has a lower abuse potential than cocaine, another inhibitor of this class. This mechanism might drive ibogaine’s effect on mood and psychological performance.
Finally, the nicotinic acetylcholine receptor activity is perhaps most likely accounting for the anti-addictive property of ibogaine. Ibogaine is a non-competitive antagonist at several receptor subtypes, most notably the alpha 3 beta 4. This receptor is an important part of reward pathways. Blocking it can dampen dopaminergic activity and reduce self-administration of various drugs.
Beyond this, ibogaine also induces upregulation of GDNF. This is a crucial neurotrophic factor that promotes survival and plasticity of neurons, amongst others. This effect likely drives the attenuation of drug craving and use by ibogaine.
What is the Evidence for Ibogaine?
Now we’ve seen that ibogaine bridges several different classes of psychoactive substances. This translates into promising clinical efficacy, particularly in substance-use disorder. Most ibogaine studies lack rigorous clinical study design – however, there are good data in opioid and cocaine craving.
Let’s briefly check out the largest study, comparing self-reported mood and drug craving measures of opiod or cocaine dependent patients. Strikingly, after an oral dose of ibogaine, patients reported significantly lower levels of drug craving. This is measured through a questionnaire which tests patients’ confidence in ability to quit, emotionality and other factors. In addition, depressive symptoms got better as well. These improvements continued to grow after one month follow-up, indicating potentially quite durable benefits. Many other conditions have preliminary data but we will not talk about them here.
In any case, the upsides look quite promising. What about the downsides?
Ibogaine’s complex pharmacology leads to considerable potential to generate adverse effects. In rats, high doses led to degeneration of neurons. They did not replicate this in primates, so it might be species dependent and less worrisome. High doses have also led to tremors and convulsions in rats.
Much more importantly, ibogaine can also negatively affect the cardiovascular system by prolonging the QT interval of the heart. This comes from strong inhibition of hERG potassium ion channels. These channels coordinate the heart’s beating through repolarization of cardiac neuromuscular junctions. Abnormally QT intervals increase risk of developing heart rhythms problems and even sudden cardiac death.
That’s why alarming reports of life-threatening complications associated with ibogaine have been accumulating. As you can see here, even young people with no other substance use are at risk. Due to the longevity of the metabolite Noribogaine we mentioned, cardiac adverse events may also occur several days. In some cases it can even be weeks after intake of a single dose of ibogaine.
The goal is not to test ibogaine mindlessly in dozens of conditions, potentially giving patients sudden cardiac arrests. Instead, we should explore safer, ibogaine-related molecules to unlock its therapeutic potential. This research needs to elucidate the underlying mechanisms of actions. If promising, the drugs should be translated into robust, objective clinical trials in humans.
So how can we shift the balance towards better safety at similar or even better efficacy?
Ibogaine Variants: 18-Methoxy-Coronaridine
The first attempt at this is an investigational molecule is 18-MC. 18-Methoxy-coronaridine is a modified ibogaine with an additional methoxy and methyl ester group. It is synthesized differently than ibogaine, so stay tuned for the last chemistry section.
These new functionalities impact the pharmacological profile a lot. For instance, low activity at sigma sites reduces risks of neurotoxicity, while lack of activity on serotonin transporters means that 18-MC is not hallucinogenic. Interestingly, the activity at the alpha 3 beta 4 nicotinic receptor is much lower, but 18-MC is much more selective for this sub receptor than ibogaine. So, we can see that in some cases, a lower affinity is not bad if it is more targeted.
A more complicated point is also that this table only shows binding affinity – but sometimes, an equally strong affinity expressed as Ki can have a much higher IC50 value, which reflects true inhibition. Unlike ibogaine however, 18-MC does not increase GDNF expression, the additional factor believed to be critical for neuroplasticity, so their mechanisms of action are potentially distinct. Overall, 18-MC seems to have a much narrower spectrum of actions. In theory, this drives a greater therapeutic index – meaning the effective dose is much lower than a potentially harmful dose.
Regarding cardiotoxicity, 18-MC inhibits hERG channels roughly 3- to 4-times weaker than ibogaine. It’s not fully clear whether this is enough to abolish the arrythmia and cardiac adverse events – just shortly, we will check out another analog which is even better.
The clinical fate of 18-MC is not clear either. The biotech MindMed – don’t confuse it with Mind Cure – completed a Phase 1 trial last year with a solid number of patients dosed. Initial data was positive with good tolerability and no serious adverse events. They also planned a larger proof of concept trial. However, they paused it due to financial reasons with new financing and partnering required to advance the program.
Instead, the company is focusing their efforts on the development of LSD in phase 2 for anxiety and ADHD, and MDMA pre-clinically for autism spectrum disorder. As we have seen in previous videos on this channel, these drugs might be very promising in these conditions. So, who knows – strong data could resurrect MC-18. Drop me a comment if you want an update on these programs in future!
Ibogaine Variants: Tabernanthalog
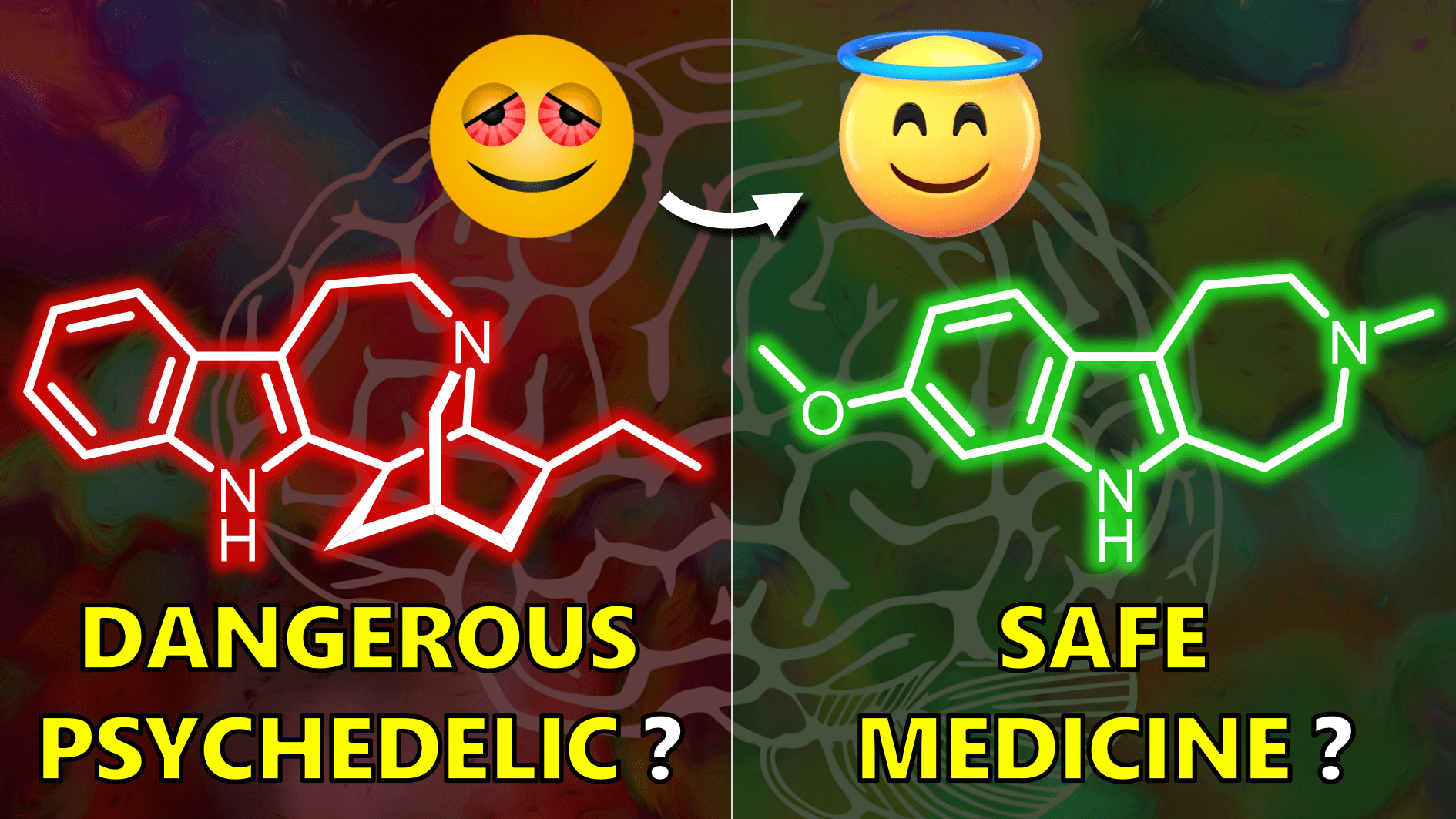
In any case, fortunately there has been a promising addition to the analog roster. A 2021 paper in Nature reported the results of another quest into ibogaine analogs. Instead of throwing more groups on ibogaine like MC-18, the logic here was to simplify ibogaine’s structure, thereby improving accessibility and elucidating which features are most important for activity. In case of the ibogaine skeleton, you can envision two different simplified ring systems – one in light green and one in blue.
Out of many compounds, the most promising is “tabernanthalog“, featuring a shifted methoxy group compared to ibogaine. Before we check out why this molecule seemed to hit the sweet spot of safety and therapeutic effect – do you have an idea how to synthesize TBG?
Even though it’s quite sizeable, it requires only one step, a Fischer indole synthesis. This reaction links this substituted phenyl hydrazine with the seven-membered ketone, creating the tricyclic TBG. The mechanism is part of many undergrad courses. The initial condensation reaction forms a phenyl hydrazone which isomerizes to the enamine form, drawn here. Upon protonation, we have a sigmatropic rearrangement which creates the C-C bond. After re-aromatization, the nucleophilic amine drives C-N bond formation via the aminal – which eliminates ammonia under acidic catalysis. We will review full syntheses of ibogaine in the final section of this video – but you can already guess that making TBG in a single step with 55% yield is infinitely easier than synthesizing ibogaine from scratch.
What are Effects Of Tabernanthalog?
First up is hallucinogenicity. While appreciated by some folks, pharmaceuticals should not elicit hallucinations. Seasoned channel viewers will recognize the classic head-twitch response assay to test for hallucinogenic potential of molecules. As a positive control, we have 5-methoxy DMT which is strongly hallucinogenic, reflected in the frequent head-banging of mice. In red is IBG – this is not ibogaine but rather the simplified version with the methoxy at a constant position. Even lower than IBG was TBG in blue with essentially no hallucinogenic potential. So, these were quite some sleepy mice instead of the energetic headbangers for 5-MeO-DMT.
Remember ibogaine’s adverse cardiac effects, mediated by the hERG channel? Both simplified analogs have much weaker inhibitors than ibogaine. The simple shift of the methoxy position between IBG to TBG comes with an additional 7-fold reduction in IC50 value. The overall 150-fold weaker binding gives TBG its promise as a quote unquote “safer ibogaine”. Obviously, this is much better than the 3-4-fold difference between ibogaine and 18-MC, the first analog we talked about.
So, safety is just one part of the equation – but does TBG also bring similar positive effects? Here is where we want to review a few interesting experiments, starting with neural plasticity.
This is the ability of neural networks to change through growth or reorganization. One way to look at it is the growth of dendrites – these are a nerve cell’s extensions which propagate electrical stimuli. Exposure of rat neurons to ibogaine, IBG or TBG all lead to more dense dendritic spines.
We can distinguish if dendritic growth is due to slower break-down of spines, or instead by a higher rate of formation. Both DOI and TBG drive growth in the same manner – they accelerate the formation of new dendritic spines.
Do these psychoplastogenic effects translate into behavioral or anti-addictive effects for TBG as well? We pointed out, there are anecdotal and initial clinical reports that ibogaine can reduce alcohol or opioid use. For this analysis, you unfortunately must make mice alcoholic by giving them the option of binge drinking. After a standard 7-weeks protocol, they compared alcohol consumption between two groups. Mice who proceeded as usual (blank) and mice who received TBG prior to the drinking session. The latter group had much lower alcohol intake both during the initial part of the consumption test, as well as acutely over the following days.
The team observed similar effects when looking at heroin as another substance with high abuse potential. Here, TBG administration also led to a much lower heroin intake – seen on the left graph – and also seeking behavior – as seen on the right graph in terms of number of lever presses during their experiment.
As a last notable effect, we look at TBG’s impact on depression. We can investigate this through a “forced swim test”. “Less depressed” mice will spend more time in motion, somewhat reflecting their drive and will to live. Even though also quite controversial, all marketed antidepressants increase swimming time in the FST – so the test is legit. The researchers performed two tests – one 24 hours after administration of TBG, and a second after one week of rest. This time, the blank positive control bar is ketamine, an effective anti-depressant. During the first test, both ketamine and TBG reduced immobility. Adding ketanserin once again abolished the effect as you can see in red. Interestingly, ketamine’s effects seemed more durable, as it still led to significant lower immobility one week after administration. TBG on the other hand looked more like the vehicle control.
We discussed previously that ibogaine and its metabolite noribogaine interact with numerous biological targets. Unlike Noribogaine, TBG or IBG showed no activity at opioid receptors. Perhaps, the higher selectivity could lead to a better drug profile down the line. On the other hand, the control experiments which ketanserin already showed us that serotonin receptors are vital for TBG’s activity. A more detailed screening revealed that TBG is both an agonist of the serotonin 2A receptor – but also an antagonist to the serotonin 2B receptor. Drop a comment if you need some more explanations on how to read these charts. The interesting thing here is that many 2A agonists are also 2B agonists, which can lead to side effects like heart valve disease. 5-methoxy DMT is a key example – as you can see in the orange plot, it inhibits both receptors in a similar manner.
Outlook on Tabernanthalog
This case study was rather simple on the synthetic design part of things. Still, I think it’s really fascinating that TBG looks like ibogaine but seems to behave differently mechanistically. Although much work on translational science into humans and dosing optimization is required, TBG might be able to overcome ibogaine’s safety limitations and unlock the potential of this class of drugs.
And last, a brief note on Mindcure. This biotech company was pursuing the development of ibogaine, garnering some attention from professional and private investors such as the chap we saw during the intro. They supposedly were on track to have fully synthetic GLP supply of ibogaine ready by end of last year – but ironically, just two weeks after, reported the result of a strategic review – the discontinuation of all activities. The psychedelic pharmaceutical market can be quite volatile, and funding challenges in recent years have definitely not helped these companies either.
As a random side note, their website was dubious from the start as they didn’t get the molecular structure of ibogaine right – unless they were showing some other analog which I missed.
So – all in all, there are some promising evolutions, but progress is sluggish. I expect that we are still far away from regulatory approvals. Instead, emerging clinics in countries where ibogaine is legal will continue to draw visits from abroad. They might be helpful for some individuals as a last resort but come at a risk of sketchy medical practices and questionable patient safety.
On the positive side, we do see increased state and federal interest in ibogaine due to the opioid problem, and psychedelics more broadly. For instance, the state of Kentucky is currently considering the allocation of 42 million dollars for ibogaine research. Out of a much bigger pocket of almost a billion in settlement funds, this looks like money well spent on larger and broader clinical trials.
Organic Chemistry: Retrosynthesis of Ibogaine
Now we will discuss not one or two, but three different approaches towards the ibogaine framework – as well as the synthesis of 18-MC.
From a retrosynthetic perspective, given the high complexity of the ibogaine scaffold, there are various disconnections that lead to sensible synthetic approaches. A quite straight-forward option uses a Fischer indole synthesis with a simpler ketone. However, most approaches include the indole from the start to guide the synthesis. One method we will review uses transition metal catalysis, while others harness the electrophilic reactivity of the indole. The gram-scale synthesis we will look at uses yet another approach based on nucleophilic substitution at the aliphatic nitrogen. Note that these syntheses focus on ibogamine – which is ibogaine lacking the methoxy group – because it is not a controlled substance.
First total synthesis of ibogamine (Büchi)
Let’s start with the pioneering first total synthesis of ibogamine, published by Büchi in 1965. It started from this pyridinium salt, which was reduced to the diene. This prepared the Diels-Alder reaction with methyl vinyl ketone which nicely builds the iso-quinuclidine core of ibogamine. Next, some redox and functional group interconversions produce the following intermediate. There are quite a few things going on, so we won’t go into it in detail – but this is a nice exercise for motivated viewers. Now, hydrogenation of the benzyl protecting group released the nucleophilic amine, which was coupled to this indole, bearing an acyl chloride.
The next task was to create the central C-C bond to connect the rings. This was achieved in two steps – under acidic conditions, the indole electrophilically attacks the adjacent ketone, and the resulting adduct was reduced with Zinc and acid. A few more steps were needed to get all ducks in order. First, a reduction removed the acetate protecting group and partially reduced the amide. To get to the fully aliphatic amine, they had to take a detour due to the reactivity of the system. Elimination of the hydroxy group with base temporarily cleaved the isoquinuclidine ring.
The link was regenerated by reduction with Zinc, which is mediated through the unsaturated ketone. Finally, a Wolff-Kishner reduction with hydrazine removed the ketone and gave ibogamine. All in all, not bad for 1965, but can we make this more efficient?
Modern Synthesis of Ibogaine
That’s exactly what the second synthesis is about. It starts off with a Palladium-catalysed heteroannulation to forge a highly functionalized indole. You will note that the ring contains a methoxy group, so this is indeed a synthesis of proper ibogaine. Next, two iodide groups were introduced – first at the indole by treatment with electrophilic NIS, and second at the aliphatic position by deprotection and SN2.
This reactive iodide should remind you of the acyl chloride we saw in the 1965 synthesis – again, it allows the introduction of the isoquinuclidine through another substitution. This can be made in a similar fashion as we saw in the 1965 synthesis as well – so these syntheses have some parallels. Interestingly, the authors noted that when using potassium carbonate as a base, there was a significant degree of intramolecular cyclization to the cyclopropane. This could be suppressed by using caesium carbonate instead. Finally, the indole and isoquinuclidine were bridged through a reductive Heck coupling, which after elimination already gave our product ibogaine. This synthesis is definitely more efficient and direct – but is there anything cooler?
Large-Scale Synthesis of Ibogaine
Three time’s a charm today. Quite recently, a paper described the gram-scale synthesis of ibogamine in just nine steps and an impressive 24% overall yield. Most notably, this approach would provide ample material to pursue even more synthetic analogs, particularly ones than are more complex than TBG.
The synthesis started from this vinylogous ester. First, a simple silylation protected the primary alcohol. Then, a Stork-Danheiser transposition with the Grignard reagent formed an enone, now bearing the ethyl group present in ibogamine. Through a Mitsunobu coupling, this fragment was linked to an indole bearing an amine. So, this contrasts with the previous syntheses where we had an electrophilic indole partner, this one is nucleophilic.
The ketone was then selectively reduced under Luche conditions and acetylated to create an activated allylic system. This set the stage for pivotal Friedel-Crafts reaction – which as we’ve seen can be mediated by Bronsted or Lewis acids. After some screening, the chemists found decent conditions with catalytic camphorsulfuric acid and lithium perchlorate at a 5M concentration. This meant that the scale-up would require massive amounts of perchlorate. Initial optimization attempts were not fruitful, as they either had to keep the quantity of perchlorate or dilute the mixture to unpractical 0.001M. Ultimately, after trying enough conditions, they got good conditions employing only 2 equivalents of magnesium perchlorate.
Finally, the only thing left was the formation of the C-N bond on the isoquinuclidine – you might remember that we highlighted this as a key retrosynthetic disconnection at the start. First, the double bond which remained from the enone was hydroborated and activated as a mesylate. And last, the nitrogen was deprotected which triggered the intramolecular SN2 reaction to give ibogamine. The whole exercise delivered 1.1g of pure ibogamine in one go.
Synthesis of 18-Methoxycoronaridine
To conclude our journey, let’s check out the initial synthesis of 18-MC starting from tryptamine – as you can imagine, it will be more complex than the ibogaine syntheses given the two additional functional groups.
The route starts off with a condensation of tryptamine to the ketone of this fragment. If you are still awake, you will notice that this ester group ultimately ends up in 18-MC. Due to the alpha-chloro group, the product can undergo an intramolecular substitution, creating a transient aziridine, and rearrange to the expanded 7-membered ring. Then, the double bond can be reduced, and the nitrogen protected.
The unique thing about this system is that upon heating, a retro 1-4 addition can fragment the ring, liberating the free amine and the alpha, beta unsaturated ester. Why is this helpful? Well, by condensing the amine with this aldehyde, a dearomative Diels-Alder reaction can be triggered. As a side note – that it really matters down the line – note that because the intermediary (e)-enamine was preferred, the product has the substituents in trans positions. Also, the newly introduced piece features the methoxy group we want to have in 18-MC.
So now, it’s all about linking up the rings properly. First, a conjugate reduction regenerates the aromatic indole and releases the quaternary carbon. Next, a hydrogenation unveils the amine, which upon deprotection of the aldehyde forms yet another cyclic enamine.
Redrawing this structure, we realize that just a final ring closure is needed to create 18-MC. This was achieved by simply heating in toluene because the additional ester group proved quite handy. It’s likely that an intramolecular proton shift facilitates formation of an anion and iminium, which can react to create the quaternary centre and deliver 18-MC. This first synthesis from 2001 did seem a bit random – and there are more efficient routes that are more analogous to ibogamine – but I thought it was nice that they used the additional ester to guide the approach.
This concludes our ibogaine journey. I hope you learned several new interdisciplinary science facts today!
References on Ibogaine, Ibogamine, Tabernanthalog & 18-MC
DARK Classics in Chemical Neuroscience: Ibogaine | ACS Chem. Neurosci. 2018, 9, 2475
Phytochemical characterization of Tabernanthe iboga root bark and its effects on dysfunctional metabolism and cognitive performance in high-fat-fed C57BL/6J mice
A systematic literature review of clinical trials and therapeutic applications of ibogaine | Journal of Substance Abuse Treatment 2022, 138, 108717
Ibogaine Detoxification Transitions Opioid and Cocaine Abusers Between Dependence and Abstinence: Clinical Observations and Treatment Outcomes| Front. Pharmacol., Sec. Neuropharmacology 2018, 9: 00529
The Anti-Addiction Drug Ibogaine and the Heart: A Delicate Relation | Molecules 2015, 20, 2208
18-Methoxycoronaridine (18-MC) and Ibogaine: Comparison of Antiaddictive Efficacy, Toxicity, and Mechanisms of Action Annals of New York Academy of Sciences 2000, 914, 369
A non-hallucinogenic psychedelic analogue with therapeutic potential | Nature 2021, 589, 474
The Total Synthesis of (±)-Ibogamine and of (±)-Epiibogamine | JACS 1965, 87, 2073
Total synthesis of ibogaine, epiibogaine and their analogues | Tetrahedron 2012, 68, 7155
Gram-Scale Total Synthesis of (±)-Ibogamine | Org Lett 2023, 25, 4567
Chemical Synthesis and Biological Evaluation of 18-Methoxycoronaridine (18-MC) as a Potential Anti-addictive Agent | Current Med Chem CNS Agents 2001, 1, 113